三重态能量转移使分子间[2π 2σ]-光化学环加成
文章出处:Roman Kleinmans, Tobias Pinkert, Subhabrata Dutta, Tiffany O. Paulisch, Hyeyun Keum, Constantin G. Daniliuc, Frank Glorius. Intermolecular [2π 2σ]-photocycloaddition enabled by triplet energy transfer. Nature 2022, 605, 477-482.
摘要:一个多世纪以来,光化学[2 2]-环加成被合成化学家用来制造环丁烷,四元碳基环。在这个反应中,通常是两个烯烃亚基(每个烯烃有两个π电子)形成两个新的C-C σ-键。近一个世纪以来,光化学[2 2]-环加合物的研究取得了巨大的进展,但目前的研究主要集中在[2π 2π]-体系中,两个π-键转化为两个新的σ-键。本文报道了以双环[1.1.0]丁烷为2σ电子的分子间[2 2]-光化学环加成反应。这种应变释放驱动的[2π 2σ]-光环加成反应是通过可见光介导的三重态能量转移催化实现的。本研究公开了一种由杂环烯烃偶联伙伴(即香豆素、黄酮和吲哚)简单、模块化和非对映选择性合成双环[2.1.1]己烷的方法。鉴于双环[2.1.1]正己烷作为生物同异构物的重要性日益增加,它们在药物研究中具有与其所替代的生物性质相似的生物性质,并且考虑到它们的获取途径有限,仍然需要新的合成方法。应用这一策略,作者可以将分子间[2 2]-光化学环加合物扩展到σ-键,并提供以前无法获得的结构基序。
[2 2]-光化学环加成物属于最基本的有机转化,是直接合成应变的环丁烷的现成工具。以完美的原子经济,一步形成两个新的C-C σ-键和多达四个立体中心,使这一过程极具吸引力。最早报道的光化学[2 2]-环加成(一种在阳光照射下介导的固体光二聚作用)是Liebermann在1877年发表的。图1a描述了光化学[2 2]-环加成物历史中其它选定的里程碑,包括分子内和对映选择性变异。特别是分子间交叉选择协议的发展受到了极大的关注。与分子内或二聚化方法相比,这些转化允许烯烃组分的模块化变化,并可快速获取分子复杂性。交叉选择性光化学[2 2]-环加成的一般概念包括选择性活化烯烃A,然后烯烃A可以与烯烃B反应(图1b)。从力学的角度来看,在这方面最相关的策略是激发一个底物进入它的三重态(T1)。这主要是因为这种三重态激发态的寿命相对较长,导致分子间产生相互作用的可能性增加。底物的三重态可以通过直接激发和激发单重态(S1)的后续系统间交叉达到,也可以通过由合适的光敏剂的三重态能量转移(ENT)介导的间接布居达到。直接激发的一个主要缺点是,大多数有机底物需要苛刻的电离紫外线(UV)照射,这经常导致不希望的和竞争性的副反应。因此,合成界最近关注于使用较温和的可见光使ENT过程克服这些选择性和激活问题。4
受20世纪60年代Prinzbach课题组关于三环系统分子内价[2π 2σ]光异构化的开创性报告的启发(图1c),作者质疑应变释放方法是否能够实现分子间[2π 2σ]-光化学环加成,其中处于三重态的激发态π-键与σ-键反应形成两个新的C-C σ-键(图1d)。
作者设想,双环[1.1.0]丁烷(BCBs)将是作者提出的[2π 2σ]-光环加成的理想选择,因为它们可以使双环[2.1.1]己烷(BCH)通过与烯烃组分的简单反应得到产物。具有结构受限的富含C(sp3)骨架的三维结构,如BCHs,在生物等立体结构中越来越重要,因此在药物化学中具有极大的价值。然而,缺乏对这些框架的实际综合访问,特别是对于高度替代的例子,固有地限制了它们在药物发现中的应用。因此,开发新的方法来实现它们的简单和模块化合成是合成界的最高兴趣。考虑到这一点,作者发起调查评估合适的烯烃偶联配体结合二苄基氨基取代BCB (化合物2a)。其中,使用蓝色发光二极管(LEDs)的可见光照射,溶剂为乙腈,三联体光敏剂为[Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (化合物Ir-F,三重激发态的能量ET = 61.8 kcal·mol-1)。幸运的是,当使用香豆素(化合物1a,ET = 62.1 kcal·mol-1)作为偶联配体时,形成期望的[2π 2σ]-环加成产物3a。使用简单的硫杂蒽酮三重态敏化剂(化合物TXT,ET = 65.5 kcal·mol-1)不仅提高了产率,而且使作者可以进行无金属的反应。在对照实验中,在没有光催化剂或光的情况下,没有观测到产物的形成。注意,在优化和底物范围分析期间,只观测到顺式异构体的形成。

图1
在确定了优化的反应条件后,作者继续研究底物范围(图2),从一系列不同功能化的BCBs开始。在作者的[2π 2σ]-协议中,一个附加的Weinreb酰胺(为进一步的下游修饰提供了潜在的位点)和一个吗啉酰胺是成功的底物,各自的产物3b和3c都具有高产率。此外,产物3d-3f显示,不同的烷基酯(乙基、异丁基和苄基)是合适的。芳基(化合物3g)与烷基酮(化合物3h)相容性好,产率高。BCB硼酸匹纳醇酯(Bpin)以合成有效收率转化为所需产物3i,为进一步多样化提供了许多机会。无吸电子基团的BCB也能反应,以1: 1的比例混合得到相应的产物3j。即使是1,3-二取代的BCB也是兼容的,导致BCH(化合物3k)具有高度修饰,桥头堡位置有两个相反的四碳中心。不幸的是,目前砜类化合物显示了这种方法的局限性。由于Weinreb酰胺BCB(化合物2b)产率最高,因此选择其作为进一步研究的标准底物。进行了基于反应条件的敏感性评估,以确保高水平的重现性。尽管大多数参数[温度(T)、浓度(c)、水和氧]的影响微不足道,但高的光强度(I)对最佳反应结果至关重要。
接下来,作者将注意力转向探索香豆素底物的范围。成功地测试了芳香环上有甲基取代的香豆素,甚至是有空间要求的底物,以较高的产率得到产物3l-3n。产物3o也表明,π-体系可以延伸到苯并香豆素部分。此外,各种卤代(Br、Cl和F)香豆素在不同取代模式下都有良好的耐受性(化合物3p-3t)。
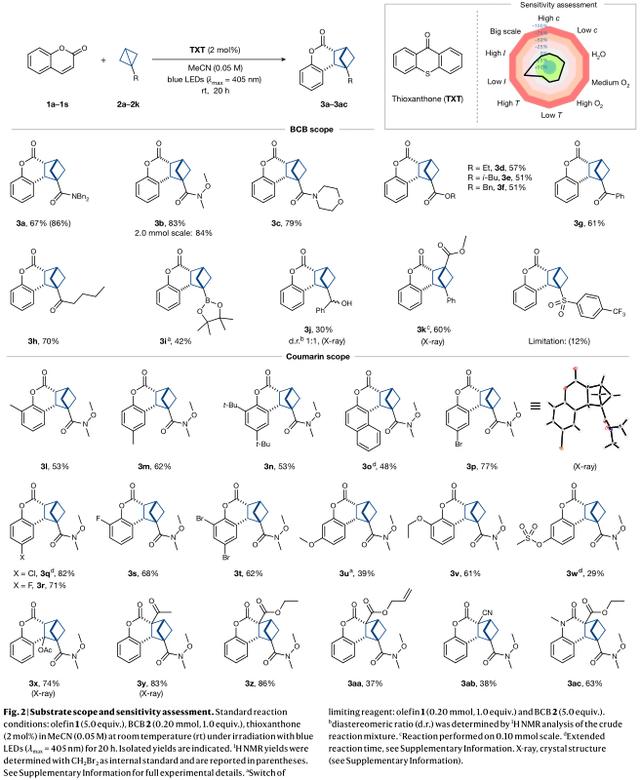
图2
此外,提供电子的烷氧基(化合物3u和3v)、甲酰基(化合物3w)和乙酰基(化合物3x)醇、酮(化合物3y)、酯(化合物3z、3aa、3ac)、系链烯烃(化合物3aa)和腈(化合物3ab)等官能团也与本协议兼容。值得注意的是,双键上的取代导致了全碳四元中心(化合物3y-3ac和3al)的构建。此外,2-喹诺酮衍生物,香豆素的氮类似物,以良好的产率得到各自的产物3ac。通过使用不同的黄酮(化合物3ad-3af)和吲哚(化合物3ag-3al)作为杂环烯烃偶联配体,进一步说明了该策略的潜力(图3a)。这些产物具有特别高的结构和官能团密度,产物3al具有三个相邻的完全取代的碳中心。
作者想知道氮杂Paternò-Büchi类型反应是否可行,作者的[2π 2σ]方法可以快速获得2-氮杂双环[2.1.1]正己烷骨架,根据取代模式,它们可以被认为是吡咯烷或脯氨酸生物同分异构体。最近,Schindler课题组报道了一个ENT激活的同2-异恶唑-3-羧酸酯分子间aza Paternò-Büchi反应。幸运的是,在作者的标准反应条件下,当使用底物4时,得到了理想的2-aza双环[2.1.1]正己烷产物5,并且产率很高(图3b)。如前所述,产物中包含的Bpin为产物多样化提供了直接的机会。因此,作为代表性的例子,产物6被氧化为相应的醇化合物7a,并用于C(sp3)-C(sp2) (化合物7b)和C(sp3)-C(sp3) (化合物7c)的偶联(图3c)。桥头堡位置的可变C-C键形成,形成四元碳中心,显示了这种策略的综合效用,因为相应的BCBs很难获得。此外,作者能够通过与哌啶(化合物8a)的氨基解和与甲醇的酯交换(化合物8b)来打开产物3b的酚内酯部分(图3d)。所得产物均为顺式产物,且产率高。这些开环实验突出了作者开发的方法的顺式选择性,因为[2 2]-光化学环加成与类似的无环烯烃的加合通常会导致对映体的混合物。最后,用过量的LiAlH4还原化合物3b,使苯酚与中间醛分子内环化为半缩醛化合物8c。

图3
接下来,作者进一步深入研究了[2π 2σ]-光化学环加成反应的机理。反应组分的紫外/可见光谱(图4a)显示,在λ = 405 nm波长附近,化合物TXT是唯一的光吸收物种,消除了标准反应条件下香豆素(化合物1a)或BCB (化合物2b)直接激发的可能性。此外,Stern-Volmer猝灭研究清楚地表明,香豆素(化合物1a)猝灭了被激发的光催化剂(这里值化合物Ir-F),而BCB (化合物2b)没有显示可检测的猝灭(图4b)。另外,用标准反应的紫外光(λmax = 365 nm)直接激发实验,反应8天后产率为14% (图4c)。考虑到只有香豆素(化合物1a)吸收这个波长范围内的光(图4a),这些实验进一步暗示香豆素(化合物1a)是激发态物种。为了排除热背景反应,在没有光催化剂的情况下,将反应混合物加热到100 oC。然而,没有观测到产物的形成(图4c)。另外,量子产率确定为Φ = 0.12 (图4d),并对一系列具有不同三重态能量的光催化剂进行了测试(图4e)。作为一个总的趋势,可以认识到产率与增加的三重态能量相关,而在氧化还原电位中没有确定的趋势。基于这些发现,并以密度泛函理论(DFT)计算为支持,图4f概述了所提出的逐步机制。
可见光激发的TXT (化合物T1)通过ENT使香豆素(化合物1a)敏化,得到的三重态香豆素使得BCB (化合物2b)形成激发复合物。这个激发复合物引起第一个C-C键形成事件,这决定了区域选择性。DFT计算表明,香豆素(化合物1a)的α-羰基位点在3位与BCB (化合物2b)结合的过渡状态在热力学上最有利(ΔΔG‡ = 2.8 kcal·mol-1),导致观测到的区域异构体。生成的1,5-二基三元体的体系间交叉允许随后的顺式选择自由基-自由基重组作为对映选择性的决定步骤,最终生成环加成产物。

图4
,




