本文来自X-MOLNews
从易得的起始原料出发,通过有机金属试剂捕获Heck C(sp3)-[M]中间体实现烯烃的区域选择性双官能化是一种快速构建复杂分子的有效方法。然而,这种方法通常会因C(sp3)-[M]物种的β-H消除得到Heck产物而受到限制。近年来,通过原位形成π-烯丙基-[M]物种和杂原子配位来稳定C(sp3)-[M]物种的策略已经成功用于抑制反应中的β-H消除过程,与此同时,自由基中间体的还原偶联也是一种烯烃双碳官能化的有效方法。

图1. 含有1,1-二芳基烷烃结构的生物活性分子。图片来源:J. Am. Chem. Soc.
乙烯基芳香化合物是最具合成价值的一类化合物,该类物种可以通过原位形成π-苄基-[M]物种来抑制β-H消除。这种烯烃双官能化的过程可用于简洁地合成1,1-二芳基烷烃,相关结构的药物分子对乳腺癌(MCF-7)、肺癌(H-460)、脑癌(SF-268)和膜蛋白FLAP(5-脂氧合酶作用蛋白)具有很好的生物活性。但是,乙烯基芳香烃双官能化的方法很少见,已发展的几例反应主要包括同二碳烷氧基化、同二芳基化/同二乙烯基化、三氟甲基化-芳基化等,还没有通用的催化方法用于乙烯基芳香化合物的三组分烷基化-芳基化来合成1,1-二芳基烷烃。
近日,美国新墨西哥大学化学系的RAmesh Giri教授课题组报道了镍催化乙烯基芳香化合物与烷基卤化物、芳基锌试剂的区域选择性烷基化-芳基化反应,由此得到1,1-二芳基烷烃产物。一、二和三级烷基卤化物均适用于该反应,不同电子特性的芳基锌试剂均可顺利参与反应。机理研究表明,反应通过Ni(0)/Ni(I)/Ni(II)的催化循环,以催化剂和烷基卤化物的单电子转移(SET)过程实现卤原子攫取。相关工作发表在J. Am. Chem. Soc. 上。
作者首先使用不同的过渡金属催化剂对2-乙烯基萘的烷基化进行探究(图2),发现在NMP中,反应以5 mol%的NiBr2作为催化剂,在室温下就能有效反应,烷基化产物2的收率为81%(entry 1)。降低环己基碘或PhZnI的用量会使产率降低(entry 2-3),使用烷基氟化物作为烷基来源不生成任何产物(entry 4)。CoCl2也能催化反应,得到中等收率(entry 7),Fe、Cu和Pd催化剂参与反应得不到任何产物(entry 8)。在没有NiBr2的情况下,反应无法发生(entry 9)。当反应在DMA中进行时得到和NMP中类似的产率,而在THF或甲苯中只有少量的产物2生成(entry 10-11)。

图2. 反应条件的优化。图片来源:J. Am. Chem. Soc.
在优化的条件下,作者考察了2-乙烯基萘与各种烷基卤化物、芳基锌试剂烷基化-芳基化反应的底物适用范围(图3)。不同一、二级烷基卤代物(I、Br)均可顺利参与反应,生成具有二、三级碳中心的产物。反应对各种官能团具有良好的兼容性,如OTBS(5)、邻苯二甲酰亚胺(6)、CF3(7-8)、OMe(4、10)和含邻位甲氧基的ArZnI(4)。

图3. 底物适用范围的考察。图片来源:J. Am. Chem. Soc.
除此之外,不同乙烯基芳香化合物也可顺利发生烷基化-芳香化反应(图4)。Cl(15-17、21、22)、OMe(19、32)、酯基(20、25)和酮羰基(30)等不同官能团可以良好地兼容。烷基卤化物中含Cl(12)、OEt(13)、OTBS(16)、烯基(17)和NCbz(24)等官能团也对反应没有明显的影响。芳基锌试剂中可含有酯基(22)和CN(29)等敏感官能团。三级烷基卤化物参与反应比一级和二级烷基卤化物和更具有挑战性。进一步的优化结果表明,(Ph3P)2NiCl2是一种很好的实现三级烷基卤化物偶联的催化剂,产物中包含季碳中心,产率良好到优秀(26~32)。作者还进一步探究了烷基化反应在药物分子衍生化(34~36)中的应用。他们以两种非甾体类抗炎药(NSAID)吲哚美辛和甲苯美辛作为母体,使用ArZnI与一级、二级卤代烷烃进行烷基化反应,以46-67%的产率得到34-36。
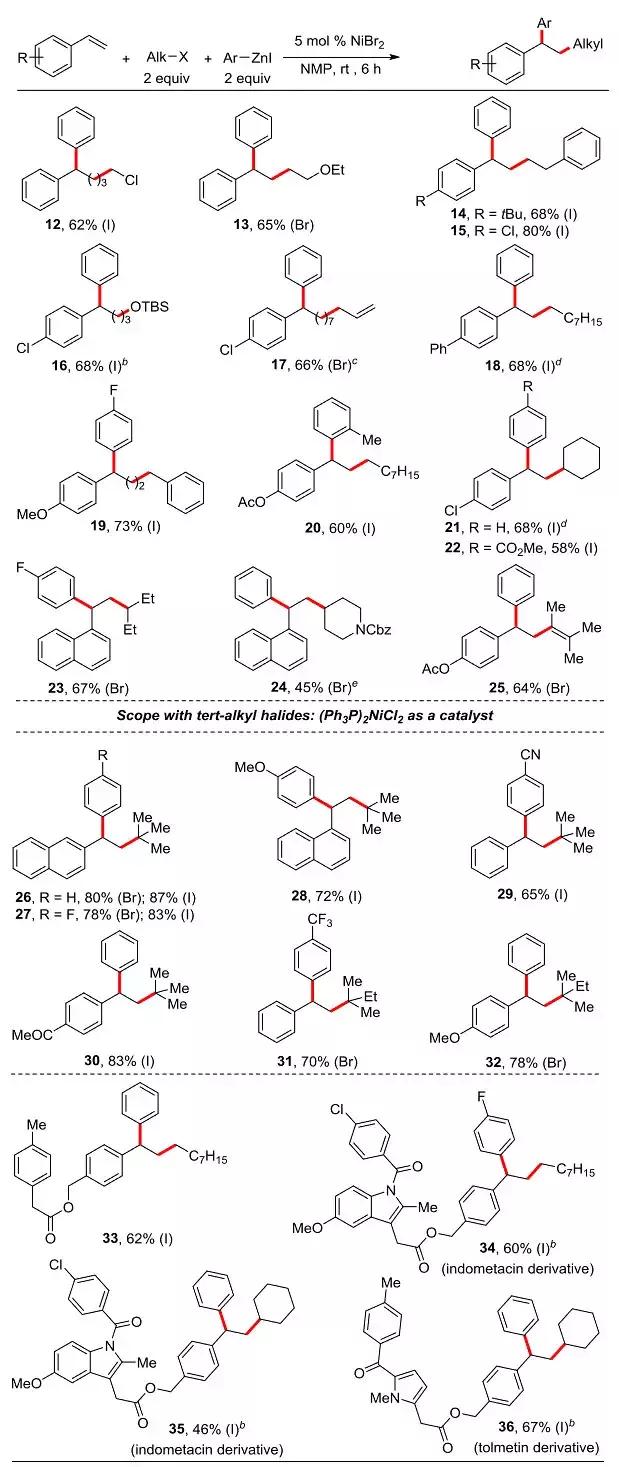
图4. 底物的进一步扩展。图片来源:J. Am. Chem. Soc.
作者还进行了机理研究以了解烷基化的过程。他们在2-乙烯基萘的存在下,将NiBr2•DME与过量的4-FC6H5ZnI混合,并用19F NMR分析了混合后瞬间形成的暗溶液,反应中4,4'-二氟联苯与NiBr2•DME的摩尔比接近化学计量比,二芳基的浓度随时间保持不变,表明在该反应条件下,NiBr2通过与ArZnI快速双转金属化和双芳基还原消除可快速还原成Ni(0)。他们还以碘代乙氧基丙烯作为自由基探针进行验证实验。在标准反应条件下,碘代乙氧基丙烯与4-氯苯乙烯及PhZnI反应,环化产物37的产率为38%。此外,当烯烃1与tBuBr和ZnI反应时,还分离到了二聚体38和预期产物26,产率分别为14%和55%,产物38来自于苄基自由基39的二聚反应。这些结果表明,乙烯基芳香化合物的烷基化-芳基化反应是通过单电子转移(SET)过程进行的。
作者还对不同烷基卤化物的反应速率进行考察,设计了竞争实验。三级烷基卤化物的反应速率快于二级烷基卤化物,而二级烷基卤化物的反应速率快于一级烷基卤化物(t-RX>s-RX>n-RX)。烷基碘的反应速率快于烷基溴,烷基溴的反应速度快于烷基氯 (RI>RBr>RCl)。这些结果与通过Ni催化剂的内层电子转移从烷基卤化物中直接攫取卤素原子的反应过程是一致的。作者还设计了4-CF3C6H4ZnI和4-MeOC6H4ZnI作为底物的竞争实验,得到相应的烷基化产物8和10,其比例接近1:1。这一结果表明,电性存在差异的ArZnI参与反应的速率没有明显差异,ArZnI试剂不太可能参与决速步骤(RDS)。

图5. 机理探究实验。图片来源:J. Am. Chem. Soc.
最后,作者进行了定量动力学研究,以确定ArZnI、烷基卤化物和乙烯在RDS中的作用(图6)。2-乙烯基萘和PhZnI在不同浓度的环己基碘化物及催化剂条件下反应,初始反应速率与环己基碘浓度和催化剂浓度呈线性关系(斜率为1.61×10-4 Ms-1;7.3×10-3 Ms-1),表明该反应对RX和催化剂均为一级反应。然而,改变PhZnI的浓度,初始反应速率没有变化,表明该反应对ArZnI呈零级反应。2-乙烯基萘对反应具有轻微的抑制作用。这些动力学研究表明,烷基卤化物直接攫取卤素原子的反应是限速步骤。

图6. 动力学实验。图片来源:J. Am. Chem. Soc.
作者提出了可能的催化循环(图7)。该反应由溶剂/烯烃稳定的Ni(0)催化剂引发,通过SET过程来还原烷基卤化物,并通过决速步卤素原子的攫取生成烷基自由基(R•)。烷基自由基与烯烃加成形成苄基自由基,苄基自由基与[NiI-X]物种重新结合,再与ArZnX进行转金属化和还原消除,生成烷基化产物,重生活性Ni(0)催化剂。

图7. 可能的反应机理。图片来源:J. Am. Chem. Soc.
总结
Ramesh Giri教授课题组发展了一种Ni催化乙烯基芳香化合物的区域选择性烷基化-芳基化反应,通过一步形成两个C(sp3)-C(sp3)和C(sp3)-C(sp2)键构建了1,1-二芳基烷烃结构。该反应对一级、二级和三级烷基卤化物以及各种芳基锌试剂均具有良好的适用性。机理研究表明,该反应是通过Ni(0)催化剂到烷基卤化物的SET过程实现卤素原子的攫取,并以此为决速步骤。
原文
Ni-Catalyzed Regioselective Alkylarylation of Vinylarenes via C(sp3)-C(sp3)/C(sp3)-C(sp2) Bond Formation and Mechanistic Studies
J. Am. Chem. Soc., 2018, 140, 9801, DOI: 10.1021/jacs.8b05374
,




