本文来自X-MOLNews
过渡金属催化是有机合成化学的前沿和重要方向,每一种金属外层电子各不相同,得失电子能力相差迥异,造就了催化的多样性和独特性,所以研究特定过渡金属并建立独特的催化反应体系对于丰富学科内涵具有重要意义。多年来,毕锡和小组注重银催化有机反应研究,开展了系统深入的研究工作,取得了一系列创新性研究结果(Acc. Chem. Res., 2020, 53, 662)。银催化化学研究的一个难点是阐明催化机理,因为有机银中间体极易发生脱银质子化,所以无法通过常规的离析关键金属有机中间体的方式来研究银催化有机反应机理。2014年,毕锡和小组报道了银催化的炔烃氢叠氮化反应,这是一类具有原始创新性的基本有机反应,为烯基叠氮化合物制备提供了非常简便的方法(Angew. Chem. Int. Ed., 2014, 53, 5305)。至今,银催化炔烃氢叠氮化方法学在国内外产生了积极影响,被同行称为“毕的银催化氢叠氮化方法学”(Bi's Ag(I) catalysed hydroazidation methodology, Donald et al. Chem. Sci., 2019, 10, 5832),逐步取代经典的Hassner方法,成为制备烯基叠氮的首选方法,有力助推了烯基叠氮化学研究。但是,这一反应存在两个关键问题亟待解决:(1)反应机理不清晰,(2)银盐催化剂用量大(20 mol%当量的Ag),导致反应缺乏实用性。
通过与张景萍教授(理论计算课题组)及长春应化所逄茂林研究员(材料科学课题组)协作研究,经过长时间探索,近期终于解决了这些问题:发现了叠氮化银(AgN3)是真正的催化物种,阐明了反应历程,建立了更加高效的叠氮化银催化的氢叠氮化反应。值得指出的是,焦宁教授和杨尚东教授等课题组曾在反应机理中提出过AgN3是可能的反应物种(Jiao et al. Angew. Chem. Int. Ed., 2013, 52, 6677;Yang et al. Org. Lett., 2013, 15, 4158; Awasthi et al. Tetrahedron Lett., 2014, 55, 1879;Bai et al. Chem. Pap., 2018, 72, 191)。作者建立的AgN3催化炔烃的氢叠氮化反应非常高效,仅需5 mol%的AgN3催化剂就能够催化这一反应,并且能够应用于较大量制备烯基叠氮化合物(30~50 mmol)。结合大量实验与理论计算研究,作者提出了AgN3催化的一种新颖的协同加成反应机制,银的类质子性使得炔与炔银间存在着可逆平衡,对于反应的发生也至关重要。这一工作开辟了银催化的有机反应机理研究的新方式,对于其它过渡金属也有一定借鉴意义。相关论文近期发表于J. Am. Chem. Soc. 期刊。

图1. 银催化炔烃氢叠氮化反应。图片来源:J. Am. Chem. Soc.
要点1. XRD分析发现AgN3是真正的催化剂
作者首先在标准条件下,通过XRD技术手段对Ag2CO3催化的反应体系中不溶性固体混合物进行了分析(图2),结果表明Ag2CO3在5 min时已经完全转化为AgN3。为了进一步证明此结果,作者进行了控制实验,发现Ag2CO3和TMSN3在DMSO条件下就能够快速的转化为AgN3,同时核磁和质谱分析其副产物为六甲基二硅氧烷(图3a)。随后,作者对该反应过程进行了密度泛函理论(DFT)计算(图3b),该反应经历了三个连续的反应过程,包括第一个Si-O键的形成、脱羧过程以及第二个Si-O键的形成,反应能垒分别为4.6、9.5和11.7 kcal/mol,因此,在该反应条件下Ag2CO3可以快速而平稳的向AgN3转化。

图2. XRD反应监测。图片来源:J. Am. Chem. Soc.

图3. (a) 控制实验;(b) M06/6-31 G(d,p)-SDD(Ag)计算水平下得到的Ag2CO3转化为AgN3的能垒图。图片来源:J. Am. Chem. Soc.
要点2. 建立了一种高效的AgN3催化的炔烃氢叠氮化反应(~ 5 mol%)
基于以上研究结果,作者进一步使用AgN3对该反应进行了条件优化(图4),他们发现5 mol%的AgN3就能够高效的催化该反应。该反应具有宽泛的底物适用范围和良好的官能团耐受性(图5);此外,该反应也可被用于天然产物结构后修饰(2al-2am)。
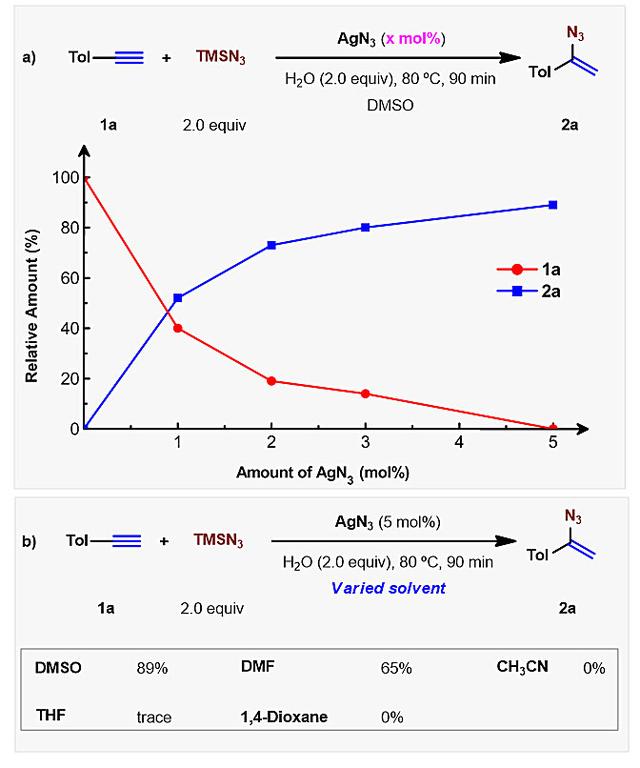
图4. 条件优化。图片来源:J. Am. Chem. Soc.

图5. 底物范围。图片来源:J. Am. Chem. Soc.
为了进一步证明该反应的实用性,作者进行了AgN3催化的放大量实验:仅使用5 mol%的AgN3就可以高效催化30-50 mmol范围内的反应,且底物范围相当宽泛(图6)。

图6. 放大量实验。图片来源:J. Am. Chem. Soc.
要点3. 结合实验和理论计算揭示一种新颖的协同加成反应机制
为了深入研究AgN3催化的反应机制,作者设计并进行了控制实验(图7)。结果表明,在反应过程中可能经历了Csp-H键的断裂产生乙炔银中间体的过程。同时发现使用乙炔银[Ag]-1b作为反应物时可以得到乙烯基叠氮化合物2b和炔烃1b的混合物,因此,反应体系可能存在端炔与炔银的可逆平衡。此外,作者还发现内炔烃作为底物时不发生反应。

图7. 控制实验。图片来源:J. Am. Chem. Soc.
随后,作者使用DFT理论计算对该反应机理进行研究。首先,作者对比考察了催化剂和底物的几种不同配位模型,发现端炔与2个AgN3分子配位形成的活性络合物A1最稳定。此外,NPA电荷分析表明,A1的C≡C三键比A1'的C≡C三键极化程度更大,更容易被活化,A1上的C2位点更容易受到银离子的攻击,这是此反应的一个重要驱动力。而内炔A1-Me的C≡C三键并不能被AgN3极化,因此银离子对A1-Me的C2原子的亲电进攻是不利的,所以内炔不能发生反应,这与实验观察到的结果是一致的(图8)。

图8. (a) 计算得到的三种络合物A1'、A1和A1-Me的构型骨架及相对自由能(kcal/mol);(b) A1的两种可能的反应模式。图片来源:J. Am. Chem. Soc.
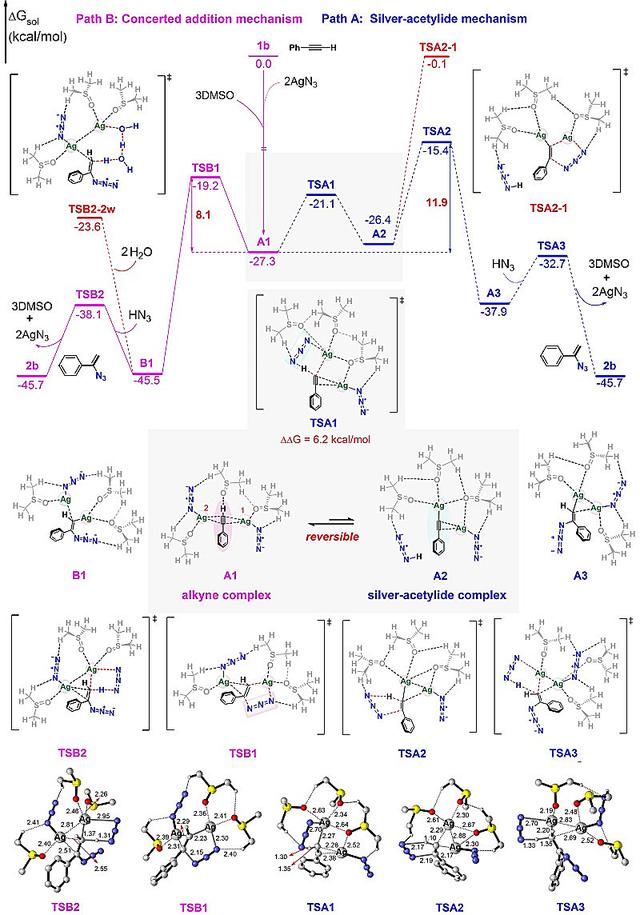
图9. M06/6-31 G(d,p)-SDD (Ag)计算水平下的AgN3催化端炔氢叠氮化反应的两种可能路径的势垒图,以及优化得到的关键过渡态结构(距离单位为Å)。图片来源:J. Am. Chem. Soc.
最后,作者对AgN3催化炔烃氢叠氮化的两种不同反应机理分别进行了探究(图9),对于经典的乙炔银机理(path A),该反应路径经历了C-H键活化,HN3对炔银的亲核加成,以及最终的脱银质子化过程,该路径中决速步骤为亲核加成TSA2,全局反应能垒为11.9 kcal/mol。另外,作者对比探究了一种新颖的协同加成反应路径(path B),该路径经历了AgN3对炔烃的协同加成和脱银质子化过程,决速步骤为协同加成过程TSB1,反应活化能垒为8.1 kcal/mol。相比于路径A,路径B的反应能垒低了3.8 kcal/mol。因此,该反应更可能是经历这种协同加成反应机理进行的。此外,作者通过NCI分析发现协同加成过渡态TSB1结构中存在额外较强的银中心与叠氮根离子之间的静电相互作用,使其结构更加稳定,具有更低的相对自由能。
总结
作者通过XRD技术研究银盐催化机理,发现了AgN3是真正的催化剂,从而发展了更加高效的AgN3催化的炔烃氢叠氮化反应,机理研究表明该反应经历了一种新颖的协同加成机制,这是AgN3首次作为催化剂应用于有机反应。本研究结果的意义不仅局限于银催化的炔烃氢叠氮化反应,而且对其它过渡金属催化机理研究也有一定启示作用。
该工作得到国家自然科学基金(21871043, 21961130376,21873018, 21471145)、吉林省科技厅(20180101185JC, 20190701012GH)和中央高校基本科研基金(2412019ZD001)项目资助。
AgN3-Catalyzed Hydroazidation of Terminal Alkynes and Mechanistic Studies
Shanshan Cao, Qinghe Ji, Huaizhi Li, Maolin Pang*, Haiyan Yuan, Jingping Zhang*, Xihe Bi*
J. Am. Chem. Soc., 2020, DOI: 10.1021/jacs.0c00836
导师介绍
毕锡和
https://www.x-mol.com/university/faculty/9495
,




