
1911年Oppenheim首次提出“变形性肌张力障碍”的概念。1984年,由“肌张力障碍医学研究基金会”提出了第一个共识定义:肌张力障碍是“一种不自主、持续性肌肉收缩引起扭曲、重复运动或姿势异常的综合征”。
随着时间的推移,早期定义和分类逐渐出现局限性。近年来,肌张力障碍定义和分类逐步得到更新,并被广泛接受。
2013年国际运动障碍协会(MDS)将肌张力定义为:一种以持续或间断性肌肉收缩引起的异常(通常是重复的)运动和/或姿势为特征的运动障碍。一般为模式化的扭曲动作,可呈震颤样。常由随意运动诱发或加重,伴有肌肉活动过溢。简而言之:过度的肌肉收缩导致的反复异常运动和/或姿势。
肌张力障碍是一种复杂的运动障碍,根据运动障碍形式不同,结合以上定义,分为相位性肌张力障碍(phasic dystonia)和强直性肌张力障碍(tonic dystonia)。
两种障碍形式常在同一个患者中共存,多以一种特征为主的混合表现。Phasic:反复扭动的状态;tonic:严重扭转、已固定不动的状态。如:痉挛性斜颈早期,头部不停地摆动,随病程延长,逐步固定地扭向一个方向。
过溢(overflow):泛化,肌张力障碍性运动累及(或激活)无关的肌肉(不在一个位置)。
肌张力障碍主要特征分类
-
发病年龄。
-
受影响的身体区域。
-
是否随时间变化。
-
是否伴随其他相关疾病。
肌张力障碍分类
主线1:临床特征
发病年龄
婴儿(出生~2岁)。
儿童(3~12岁)。
青少年(13~20岁)。
早发成人(21~40岁)。
晚发成人(>40岁)。
症状分布(身体受累区域)
局灶型:仅一个区域受累,如眼睑痉挛、口-下颌肌张力障碍、颈部肌张力障碍、喉部肌张力障碍和书写痉挛。
节段型:2个或2个以上相邻区域受累,如颅段cranial肌张力障碍和双上肢肌张力障碍。
多灶型:2个不相邻或2个以上(相邻或不相邻)区域受累。
全身型:躯干和至少2个其他部位受累。
偏身型:半侧身体受累。
时间模式(时间特征)
疾病进程:稳定型或进展型。
变异性:持续型、动作特异型、日间波动型和发作型。
伴随症状
单纯肌张力障碍或合并其他运动障碍。
单纯型:肌张力障碍是一种运动症状,可伴震颤。
复合型:合并肌阵挛或帕金森综合征。
复杂型:合并其他神经系统或全身系统疾病表现。
主线2:病因学
神经系统病理学
变性证据。
结构损害证据。
无变性或结构损害证据。
遗传性或获得性
-
遗传性
常染色体显性(DYT1/DYT5/DYT6/DYT11/HD……)。
常染色体隐性(Wilson disease/PKAN/PARK2……)。
X-相关隐形(DYT3……)。
线粒体。
-
获得性
围产期脑损伤。
感染。
药物。
中毒。
血管病。
肿瘤。
脑损伤。
精神疾病。
特发性(不明原因)
散发性。
家族性。
肌张力障碍病因
-
大多数是特发性(原发性),难以找到病因。
-
获得性肌张力障碍病因:药物、毒素、丘脑或基底节肿瘤、出血或缺血损伤、感染、炎症和围产期缺氧缺血性损伤等。
-
也有许多遗传原因:大多数为常染色体显性遗传,TOR1A(DYT1)或THAP1(DYT6)突变引起早发性全身性肌张力障碍;DYT5a-GCH1突变引发多巴反应性肌张力障碍(Segawa病、DRD);也有常染色体隐性遗传:X连锁肌张力障碍-帕金森综合征(Lubag肌张力障碍、DYT3-TAF1),常染色体隐形变异型DRD(DYT5b-th)。
-
获得确切病因诊断可能是一项挑战,但是,病因诊断很重要,其中一些有非常特异性的治疗方法。
临床常见类型
按身体区域分布
-
全身型肌张力障碍:扭转痉挛。
-
颈部肌张力障碍:痉挛性斜颈。
-
颅面肌张力障碍:眼睑痉挛、口-下颌肌张力障碍、Meige综合征(眼睑 口-下颌)、喉部肌张力障碍。
-
偏身肌张力障碍。
与随意运动或扳机点关系
-
书写痉挛。
-
音乐家肌张力障碍。
-
发作性肌张力障碍:突发出现的肌张力障碍动作,通常由某种因素诱发,可自行缓解。
发作性运动诱发的运动障碍(PKD):由突然的动作诱发,表现为短暂的自限的肌张力障碍性姿势发作。
发作性过度运动诱发的运动障碍(PED):肢体长时间运动后表现出的肌张力障碍,由跑步、游泳等持续运动诱发,长时间步行后导致足内翻。
发作性非运动诱发的运动障碍:因饮酒、饮茶、咖啡、饥饿、疲劳、情绪波动诱发。
肌张力障碍诊断步骤和流程
-
判断所患疾病是肌张力障碍的某种类型,还是类似于肌张力障碍的某种疾病。
-
详细了解病史及体格检查,根据4种临床表现特征分类。
-
考虑潜在病因:特发性(原发性)?获得性(继发性)?遗传性、基因检测。
肌张力障碍诊断
-
发病年龄:为寻找潜在病因提供依据。成年发病:特发性;儿童期发病:通常有可识别的原因。
-
症状随时间演变:为寻找潜在病因提供进一步指导。
成年发病的局灶性肌张力障碍:数周至数年内进展,以后发展较慢或难以发现。
数小时或数天内快速进展:功能性(心理性)原因,药物作用(安定剂、血管或免疫事件),遗传性疾病(快速发作的肌张力障碍-帕金森综合征)。
昼夜波动:多巴反应性肌张力障碍的特性。
突然发作:阵发性运动障碍的特征。
-
病史和体格检查:肌张力障碍是疾病的唯一表现,还是其他可识别的临床综合征的相关表现。
孤立性肌张力障碍(以往称为原发性肌张力障碍):肌张力障碍是疾病的唯一表现。
复合型肌张力障碍:肌张力障碍是更复杂的临床综合征的表现之一,如:肌张力障碍合并肌阵挛;肌张力障碍合并肝病等。
-
确定受影响的身体部位:为治疗方法提供重要指导。
肉毒素:局灶性肌张力障碍首选治疗方法。
口服药物、手术:常用于全身性肌张力障碍。
-
大多数成年发病的局灶性或节段性肌张力障碍:诊断性检查正常。
-
肌电图(EMG):显示过度的肌肉活动,通常不进行,无特异性。
-
头颅影像:偏身型、全身性肌张力障碍,存在其他神经系统体征,症状或异常快速进展,排除脑结构异常。
-
成年特发性病例:不需要基因检测。
-
有家族史或肌张力障碍与特定遗传性疾病综合征相关:基因检测。
-
大多数早发性肌张力障碍(儿童、年轻人):血液、尿液和脑脊液检测,发现病因可能性大。
-
头颅影像:可发现特定的脑结构缺陷或特定的灰质或白质疾病。
-
有家族史或肌张力障碍与特定遗传性疾病综合征相关:基因检测。
-
一般推荐儿童及青少年肌张力障碍患者做基因检测。
-
诊断明确的成年人肌张力障碍患者一般不做基因检测。
-
帮助诊断和明确病因,尽管目前基因诊断不一定对治疗有直接帮助(本身治疗手段有限),但某种程度上可指导治疗,如DBS适应证选择,是目前提倡的精准医学内涵之一。
-
一般由基因公司做肌张力障碍的成套基因检测。
-
Fahh-Marsden肌张力障碍评定量表(BFMDRS):广泛使用,评估成人、儿童、全身性、节段性肌张力障碍及DBS疗效评估。
-
多伦多西部痉挛性斜颈评定量表(TWSTRS):广泛使用的颈部肌张力障碍评定量表。
-
肌张力障碍影响躯干和至少其他2个身体部位。
-
扭转痉挛。
-
各年龄段均可发病。
-
儿童起病多见。
-
初期表现为局部灶型肌张力障碍,以后波及全身。
-
扭转运动和姿势异常。
-
2011年EFNS定义:双下肢 至少1个其他部位(通常是1个或双上肢)。
-
2013 MDS定义:躯干 至少2个其他部位。
-
痉挛性斜颈。
-
通常30~40岁发病。
-
男女比例约1:2。
-
早期常为阵发性,后可发展至持续性的斜颈,导致头位固定。
-
受累肌肉常有痛感。
-
感觉诡计:可短暂减轻异常姿势和不自主运动严重程度的主动性动作,常通过对疾病累及身体部位或邻近部位施加包括轻度触觉在内的刺激,改善症状。
-
法国神经病理学家Henri Meige在1910年首先描述。
-
中、老年女性多见。
-
眼干、畏光、瞬目、眨眼增多。
-
痉挛性闭眼。
-
不自主口周运动(张口、闭口、咧嘴等)。
-
晚期可致功能性失明,丧失独立生活能力。
-
包括:眼睑痉挛和口下颌肌张力障碍。
-
皮质-基底节-丘脑-皮质。
-
基底节受损或功能障碍。
-
直接通路和间接通路功能失衡。
-
GPi/SNr兴奋性降低,减少了对丘脑抑制,运动皮层兴奋性增强。
-
与帕金森病病理生理机制相反,但DBS治疗同样有效,因此该经典模型不能解释二者差异。
-
不同脑区参与的运动网络功能障碍。
-
多个脑区在不同类型肌张力障碍中均起重要作用。
-
皮质-基底节-小脑-丘脑-皮质。
-
基底节神经元活动时空模式改变:非人类灵长类动物模型基底节神经元放电LFP频率改变,低频振荡-时间模式改变;该区域范围内的神经元异常同步低频振荡-空间模式改变。
-
大多数与肌张力障碍相关的基因主要在纹状体和小脑中表达,这两个脑结构在肌张力障碍病理生理中的具有关键作用。
-
多巴胺能信息异常,主要涉及基底节的直接通路和间接通路,如:多巴反应性肌张力障碍、DRD。
-
异常基因→转录调节异常→内质网功能异常→Eif2α异常→突出可塑性改变。
-
不同环路水平(皮层、基底节、脑干、小脑、脊髓)抑制,兴奋剂增强→感觉运动信息整合异常→过度肌肉收缩→临床症状。
实验室检查
基因检测
肌张力障碍评定量表
全身型肌张力障碍
全身性肌张力障碍定义与以往的区别:
颈部肌张力障碍
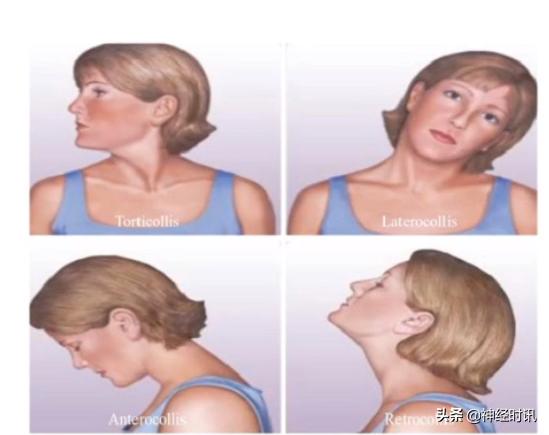
图1 颈部肌张力障碍常见的4种类型
Meige综合征
肌张力障碍机制——基底节环路

图2 经典基底节环路模型
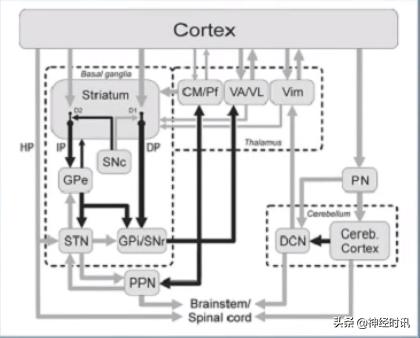
图3 肌张力障碍病理模型
病理生理学——基因及神经生物学
未完待续,下回且看肌张力障碍的治疗
编辑 | 乔婷婷
审校 | 丁慧鑫
,




