小分子酮的有机催化立体选择性氰化硅基化
文章出处:Hui Zhou, Yu Zhou, Han Yong Bae, Markus Leutzsch, Yihang Li, Chandra Kanta De, Gui-Juan Cheng, Benjamin List. Organocatalytic stereoselective cyanosilylation of small ketones. Nature 2022, 605, 84-89.
摘要:酶的立体选择性是大多数化学催化剂所无法比拟的,特别是在小分子底物的转化方面。根据“锁和钥匙理论”,酶有限域活性位点以适应其特定的反应底物,这是化学催化剂通常缺乏的特征。在这方面,一个有趣的例子是酮和HCN生成氰醇,因为这个反应可以被各种类型的催化剂催化,包括生物的、无机的和有机的催化剂。作者现在报道广泛适用的限域有机催化剂的发展高度对映选择性的氰化硅芳酮和脂肪族酮,包括挑战性的2-丁酮。其药物相关产品的选择性(98: 2的对映体比)是任何其它催化剂类,包括工程生物催化剂所不能比拟的。作者的结果表明,在转换小分子的,无偏倚的底物时,限域的化学催化剂可以设计,且仍然能在广泛的底物范围内提供高选择性。
酶在其底物上工作时所显示的辨别能力是独特的。通常,只有一个底物被接受。酶具有的区域选择性和对映选择性为化学家创造完美催化剂提供了灵感。当涉及到小分子底物时,这种探索尤其艰巨,因为在对映选择性合成过程中,小分子底物的处理是出了名的困难。这一类的一个例子涉及2-丁酮的对映选择性催化。例如,考虑到酮取代基的空间位阻体与其对应的A值(甲基为1.74、乙基为1.75)相似,这样的立体选择性过程是一项艰巨的任务(图1a)。虽然酶和Ir配合物已被描述以高对映选择性将2-丁酮还原成相应的醇,但亲碳核试剂的亲核加成通常效果不佳。这也适用于在2-丁酮中加入HCN生成氰醇的合成。值得注意的是,这种氰醇的水解产物,2-羟基-2-甲基丁酸,是三种上市药物苄氯贝特、克利贝特和甲乙双酮中含有的特权药效基团。此外,(S)-对映体用于制备COX-2抑制剂和PPAR激动剂,而(R)-对映体构建了几种具有生物活性的天然产物的骨架或侧链。据作者所知,到目前为止,只有羟腈裂解酶(HNL)在2-丁酮的不对称氢氰化反应中取得了令人满意的结果,当使用工程酶LuHNL时,最大对映选择性为93.5: 6.5 (87%的对映选择性)。先前开发的手性硫脲有机催化剂和手性salen钛配合物对这种重要的底物只产生少量的对映体控制(图1b)。作者受到最近研究的启发,使用强和限域的酸催化剂来控制困难的底物和反应。例如,作者已经证明:在这种限域的亚胺二磷酸酰酯(IDPi)催化剂的存在下,乙醛烯醇硅烷只与另一种醛以高对映选择性发生反应。作者对Diels-Alder和氢芳基化反应的研究进一步鼓舞了自身,作者注意到IDPi催化剂可以区分乙基和甲基,尽管具有适度的对映选择性。这些结果表明了限域催化剂可以显示的控制,以及硅离子不对称反阴离子定向催化(Si-ACDC)的潜在催化原理,作者推断,一种定制的、强的、可能甚至更限域的硅烷IDPi有机催化剂可以完成2-丁酮的高度对映选择性氰化硅化(图1c)。作者认为,一个临界硅基氧碳苯阳离子可能与其限域的IDPi反阴离子以这样的方式配对,两个对映体中只有一个会暴露于氰化物的亲核试剂的攻击。

图1
事实上,经过广泛的催化剂评价,催化剂IDPi 2被发现是一个特别有前途的基序,并以98:2的对映选择性及近似100%的产率提供产物6,这是2-丁酮获得的最高的对映选择性。此外,IDPis 3-5也是一种特殊的催化剂。使用催化剂2-5,探索了酮的作用域(图2)。一开始就对含甲基和长烷基的脂肪族酮进行了测试。作者的IDPi催化剂性能良好,产率为90-97%,对映选择性在93: 7-98: 2之间,与烷基链的长度无关(产物6-12)。氯代酮也被发现是合适的底物,相应的硅基氰基丙烷化合物13和14分别以94%和95%的产率得到,对映选择性为95: 5。带有烯基取代基的底物也具有良好的耐受性,其产物15的对映选择性为92.5: 7.5,产率为92%。此外,含保护羟基取代基的酮很容易在95%的产率下得到所需的产物16,对映选择性为87: 13。此外,在3,3-二甲基环己酮的反应条件下,还可以成功地使用环酮,得到产物17,其产率和对映选择性都很高。2-环己酮反应的主要产物为1,4-加成产物18,对映选择性为91: 9,产率为45%。与之相比,在适当的对映选择性下,偶联的无环酮类化合物只能得到1,2-加成产物。芳基取代的脂肪族酮的反应类似,电子效应和芳基上的取代模式都没有显著影响对映选择性(对映选择性为91: 9-99: 1) (产物19-29)。
随后,作者探索了芳香酮在苯环上不同位置的给电子基团(甲基和甲氧基)和吸电子基团(F、Cl、Br、CF3)的反应,以中等到高的产率和高的对映选择性(高达98: 2)得到所需的产物30-41,虽然需要更高的催化剂负载量。具有中等碱性杂环取代基的呋喃基和噻吩基的酮的氰基化反应也进行得很顺利,得到的产物42-44,产率适中,对映选择性高。令人高兴的是,甾体衍生物45也能以92%的产率获得,并且具有显著的对映选择性(> 95: 5)。值得注意的是,在无水条件下进行反应,在室温下进行预干燥,反应时间较短。
为了证明作者的反应的合成效用,作者进行了4-苯基丁烷-2-酮的氰基硅基化,在95: 5的对映选择性下,以95%的产率得到产物23。富含对映体的叔氰醇是重要的合成组分,易于转化为天然产物和具有生物活性或药理活性的化合物。研究了氰醇前驱体化合物23的潜在合成趋势。在简单、温和、简洁的条件下,可以在不降低对映选择性的情况下,较好地制备相应的氨基醇化合物46、游离氰醇化合物47、醛化合物48和恶唑啉化合物49。此外,作者也以克为单位生产的硅基氰醇化合物6,可以很容易有效地转化为2-羟基-2-甲基丁酸化合物50,这是合成有效抗炎药物COX-2抑制剂化合物51的重要中间体,以及目前可能作为外消旋体应用的各种对映体药物。
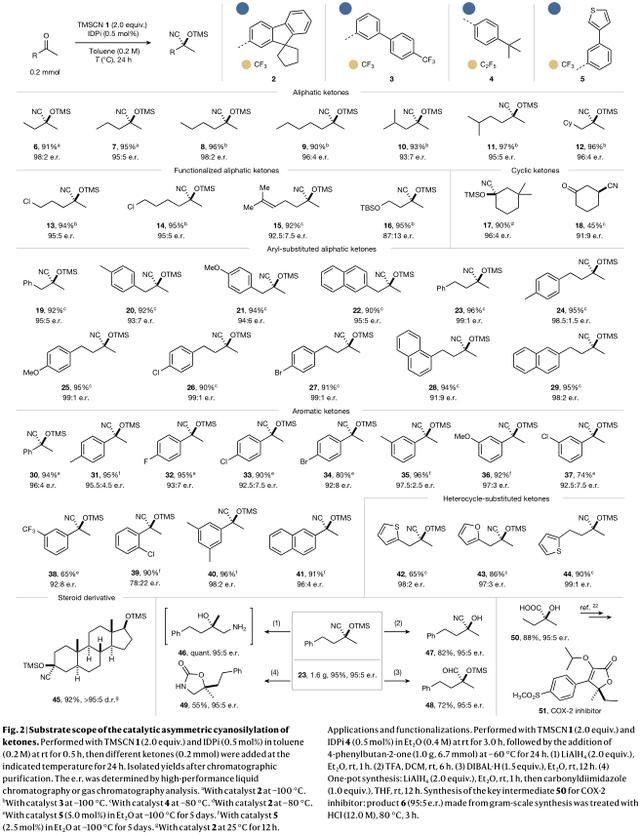
图2
根据作者之前建立的硅-氢交换反应,在二磺酰亚胺(DSI)催化下独家得到烯醇硅烷。例如,4-苯丁烷-2-酮以75%的产率和15: 1: 1比例的[(Z)-53: (E)-53: 54]生成烯醇硅烷(图3a,公式1)。相反,催化剂IDPi 4使酮化合物52进行氰基硅烷化,以92%的产率和92: 8的对映选择性得到所需的硅基氰醇产物23(图3a,公式2)。出乎意料的是,作者发现,即使在这些条件下,以IDPi 4为催化剂,1H NMR谱图也能清晰地检测出相应的烯醇硅烷化合物(Z)-53和54 (图3b)。在进一步的对照实验中,将烯醇硅烷混合物与HCN和0.5 mol.%的催化剂4直接反应,于甲苯-d8中在-80 oC下反应24小时,以70%的产率和96: 4的对映选择性得到硅基氰醇化合物23(图3a,公式3)。为了更深入地了解反应机理,作者采用1H NMR技术对反应过程进行了监测。如图3c所示,烯醇硅烷化合物54很容易与HCN反应,并在10分钟内被完全消耗。(Z)-烯醇硅烷的反应在20小时内完成,而对应的(E)-烯醇硅烷实际上不是IDPi催化硅基氰化反应生成的,在标准条件下几乎与HCN反应,导致起始物质的不完全消耗。作者还利用1H NMR获得的动力学数据,通过变量时间归一化分析对反应进行了分析(图3d)。当按照Burés所描述的过程进行时,作者发现催化剂作用下的整个反应是一级反应。

图3
采用密度泛函理论(DFT)计算方法研究了IDPi 2催化下TMSCN对2-丁酮的去原硅化和氰硅化反应的机理。如图4a所示,IDPi催化剂首先与TMSCN进行可逆的原位硅化反应,生成活性催化剂INT1 ([X-TMS][HNC],X = IDPi-),这也已被NMR检测到。值得注意的是,该过程得到的是异氰化氢(HNC),而不是HCN (ΔG≠TS0 = 11.1 kcal·mol-1对比ΔG≠TS0’ = 38.4 kcal·mol-1 (图4a)。然后,酮的底物被硅基化催化剂(ΔG≠TS1 = 12.8 kcal·mol-1)激活,形成一种氧碳苯中间体INT2,其中阳离子物种[TMS-2-丁酮] 与反阴离子[X-HNC]-配对。在IDPi的催化下,ΗΝC很容易与HCN相互作用(ΔG≠HNC→HCN = 2.4 kcal·mol-1和ΔG≠HCN→HNC = 16.3 kcal·mol-1,图4d),使INT1和INT2分别与含有HCN的对应中间体INT1’和INT2’相互作用。虽然不稳定的互变异构体HNC导致INT1和INT2的能量更高,但它比HCN更容易对氧碳苯中间体进行亲核攻击。计算得到的HNC氰化过渡态(TS2-S)的激活自由能为17.0 kcal·mol-1,远低于HCN (TS2’-S为30.0 kcal·mol-1),且在该反应条件下可以克服。一旦越过活化能垒,硅基化氰醇产物可生成并释放能量(ΔΔG(6-INT2) = -12.7 kcal·mol-1)。在另一种途径中,烯醇硅烷产物(Z)-53很容易通过简单的去质子化过程(TS3)形成,这与烯醇硅烷的NMR检测结果相一致(图3a,公式2和图3b)。更重要的是,去质子化过程是内源性的、可逆的,因此烯醇硅烷可以很容易地重制为氧化碳苯中间体INT2’,用于进一步的转化(例如氰化),这与实验结果一致(图3a,公式2和公式3)。因此,计算结果支持烯醇硅烷的生成是快速和可逆的,烯醇硅烷可以重制并转化为热力学上有利的硅基氰醇产物6。
此外,作者还计算了对映选择性的起源。结果表明,TS2-R比TS2-S高3.0 kcal·mol-1,与实验观测到的对映选择性相吻合。如图4b所示,(S,S)-IDPi 2的三维结构和空间位向图显示,N-P-N-P-N键、空间要求高的Ar取代基和Tf基团构成了一个局限的、深的手性口袋(埋藏体积百分比高达73.4%)。在导致R和S产物的最低能量跃迁态(TSs)中,羰基碳鎓中间体以两种不同的方向存在于手性口袋中。在TS2-S中,小甲基群位于西南象限,TMS群位于受阻较少的中心区域,沿z轴向外(图4c,左),而TS2-R将庞大的TMS群定位在拥挤的西南象限(图4c,右),这将扭曲TMS,破坏其与羰基的共面排列(α(Me-C-O-Si) = -40o)。因此,氧和羰基碳的p轨道重叠减少,与TS2-S相比能量更高,为1.6 kcal·mol-1。因此,作者推测氰化的对映调控可能是由于IDPi的高度受限结构和空间偏倚。进一步的DFT研究也证实了反应结果对IDPi催化剂埋藏体积百分比的依赖性。
基于积累的实验、光谱和计算数据,作者现在可以提出IDPi催化剂使酮的氰硅化的机理。因此,催化剂与TMSCN的初始硅基化生成硅基化催化剂,进而激活酮提供硅氧基碳烯中间体,在反应条件下,该中间体可以很容易地被催化剂的手性阴离子去质子化,从而提供相应的烯醇硅烷(图4d)。作者认为这一过程只是一个非循环现象,因为烯醇硅烷可以很容易地被酸性IDPi物种重新制备,提供羰基碳鎓中间体,然后被HNC捕获提供最终产品,同时再生IDPi催化剂。整个过程的特点是:由Si-H交换反应生成动力学上有利的烯醇硅烷,由硅氧碳铵离子与HNC反应生成热力学上有利的硅基氰醇。

图4
总之,作者证明了适当设计的手性和限域酸可以催化芳香族和脂肪族酮的不对称氰基硅基化反应,包括极具挑战性的2-丁酮。作者的工作可以作为一种鼓励,鼓励化学家创造出与酶相媲美的卓越的、有时甚至是极端选择性的催化剂。作者也期望自己的方法可以用于天然产物的合成和药物。
,




