本文来自X-MOLNews
电催化反应可以避免在氧化还原反应过程中使用成本较高的化学试剂,减少不必要的副产物产生。近年来,过渡金属催化与电化学合成结合实现C-H键的活化反应取得了相当大的进展。Pd催化体系在加热条件下结合电化学手段实现以上过程已发展得较为成熟。2017年,德国哥廷根大学化学系的Lutz Ackermann教授课题组发展了廉价的Co催化体系,在不加入其它氧化剂的情况下实现了电化学氧化的C-H键活化过程,芳基甲酰胺与端炔可以发生步骤经济性的[4 2]环化反应。基于对C-H键烷氧基化反应的机理研究,作者阐明了C-H/N-H键活化的过程,但炔类底物还局限于端炔,发展内炔参与的反应仍然非常重要。

图1. 端炔参与的[4 2]环化反应
近日,该课题组又在J. Am. Chem. Soc.上发表文章,仍旧以钴盐作为催化剂,通过电化学氧化的C-H/N-H键活化实现了芳基甲酰肼对内炔的[4 2]环化反应。电化学氧化C-H键活化可以在室温的反应条件下进行,H2是唯一的副产物。该反应成功的关键在于N-2-吡啶基肼结构的引入。作者还在室温条件下成功通过电化学还原、钐催化的裂解过程消除产物中辅基。
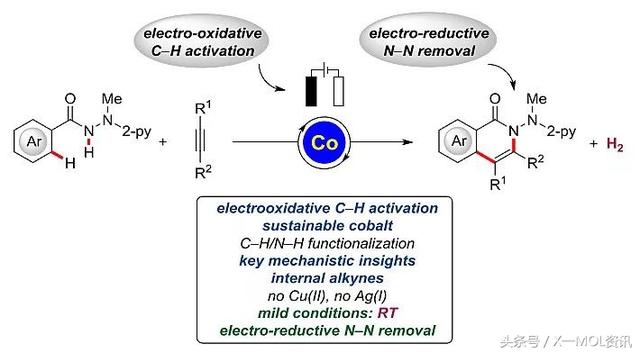
图2. 电化学氧化、钴催化的C-H/N-H键活化反应
作者首先探索了2-吡啶-苯甲酰肼1a对内炔2a电化学氧化[4 2]环化的反应条件。经过大量的条件筛选,作者发现以TFE作为溶剂,NaOPiv作为添加剂(entry 1-4)可以得到预期的电化学氧化C-H键活化的产物。反应温度为23 °C(entry 4-6)时,C-H/N-H键即可发生有效的活化。当丙戊酸作为添加剂(entry 7-11)时,体系的催化性能可进一步提高,体系中加入特戊酸作为添加剂可以提高底物溶的解度和化学选择性,并实现更有效的阴极还原,使催化性能得到进一步改善。

图3. 反应条件的筛选
在此基础上,作者还探讨了苯甲酰胺底物中N-取代基在反应中的作用。经常使用的N导向基团,如8-喹啉基或2-N-氧吡啶基取代基都不能使内炔2a发生转化,仅2-吡啶酰肼1a可得到相应的C-H/N-H键活化产物。

图4. N-取代基在反应中的作用
有了最佳的反应条件,作者对内炔底物的适用范围进行了考察。对于二取代的乙炔,取代基可以是修饰给电子或者吸电子基团的芳基、烷基,甚至酯基,反应都可以顺利转化得到相应的[4 2]环化产物。电化学氧化的C-H/N-H键活化在室温条件下就可以有效地发生。NMR实验研究表明,非对称取代的炔烃2g-2i的环化具有良好的区域选择性(图5b)。该反应还适用于芳香烃1b-1m的位点选择性官能化(图5c)。常见的亲电官能团都可以兼容,包括酯基、氯、溴、碘和氰基取代基,这对于产物的进一步多样性衍生化是非常有价值的。


图5. 内炔底物适用范围的考察
这种电化学氧化的C-H/N-H键活化过程不仅仅限于内炔烃2,各种末端炔烃7也同样适用(图6)。许多芳基取代的炔烃也是合适的底物。对氧化条件敏感的噻吩结构也能很好地兼容,氢位于氮的远端时反应具有极好的区域选择性。空间位阻大的叔丁基炔7q也是理想的底物,烷基腈7r也可顺利参与反应,没有亲核进攻的副反应发生。该反应还可以进一步实现厄洛替尼(erlotinib)衍生物8at的后期多样化转化。

图6. 端炔底物适用范围的考察
作者还对反应的机理进行了探究。分子间的竞争实验表明富电子芳香烃1参与反应更具有优势,该结果与协同的金属化-去质子化竞争实验并不一致,但可以通过碱辅助的内亲电取代(BIES)来合理解释。反应以同位素标记的D2O作为共溶剂并没有受到任何影响。作者还设计了C-H键活化的动力学研究,其动力学同位素效应(KIE)KH/KD≈1.1。气相色谱分析证实了H2是该反应唯一的副产物。

图7. 机理探究实验
作者还利用质谱法探究电化学C-H键活化的关键中间体(图8)。电喷雾电离(ESI)分析为七元钴(III)杂环的形成提供了有力的支持。

图8. 质谱分析
对于催化剂的电化学作用方式,作者通过循环伏安研究揭示了Co(OAc)2、NaOPiv和1a在MeOH中形成钴(Ⅲ)物种的过程,发现了新的宽氧化波,其起始电位为0.52 VSCE,电流最大值为0.99 VSCE。以上结果表明,底物存在时,钴(II)氧化为钴(III),电位大大降低。而在混合体系中添加炔7a并不会导致反应明显的淬灭。

图9. 100 mV/s时MeOH体系中的循环伏安曲线
计算结果表明,该反应的区域选择在很大程度上是由吡啶的杂芳基与芳基乙炔单元之间的次级π-π相互作用决定的。

图10, 关于区域选择性的计算结果
此外,缺电子的吡啶基和富电子的芳基与非对称的炔发生C-H/N-H键活化的计算结果进一步支持了这种π-π相互作用。

图11. 非对称的炔2k迁移插入时区域选择性的计算结果
作者在电喷雾质谱分析中已观察到迁移插入得到的七元钴(III)杂环9(图12)。该物种在三重态状态下发生简单的还原消除,而没有发生自旋转换,由此形成钴(I)物种,为阳极氧化再生催化活性物种奠定了基础。同时,作者还发现弱色散相互作用能稳定临界过渡态。

图12. 钴催化内炔参与的[4 2]环化反应的计算结果
基于以上实验和计算结果,作者提出了可能的反应机理。电化学氧化形成的具有催化活性的钴(III)羧酸盐物种10通过阳极氧化启动反应,随后通过羧酸酯辅助的C-H键活化得到中间体11,迁移插入产生七元钴(III)杂环物种9。9还原消除得到预期的产物3,并形成钴(I)物种12,阳极氧化再生催化活性的钴(III)羧酸配合物10。整个过程未使用有毒和昂贵的金属作为化学计量的氧化剂,生成的H2是该反应唯一的副产物。

图13. 可能的催化循环
最后,作者还尝试了C-H键活化反应中第一例电化学消除导向基团的方法。他们在室温条件下通过电化学还原的方式,以DMF作为溶剂,n-Bu4NPF6和NaI等摩尔混合作为电解质,23 ℃下使用镁阳极在催化量的SmI2存在下即可得到目标产物13cc,收率为90%。

图14. 电化学还原的方法消除酰肼
总结
Lutz Ackermann教授课题组报道了钴催化内炔参与的[4 2]环化反应,实现了电化学氧化的C-H/N-H键活化。该反应可以在温和的条件下进行,原料和催化剂来源丰富,并具有良好的区域选择性,避免了过渡金属氧化剂的使用,产生的H2是唯一的副产物。详细的机理研究、质谱分析和DFT计算表明以区域选择性的方式形成关键的七元钴(Ⅲ)杂环物种至关重要。该方法对含氮杂环的合成以及药物的后期衍生化具有非常重要的应用价值。
原文
Electroremovable Traceless Hydrazides for Cobalt-Catalyzed Electro-Oxidative C–H/N–H Activation with Internal Alkynes
J. Am. Chem. Soc., 2018, 140, 7913, DOI: 10.1021/jacs.8b03521
,




