
01
导读
C-H 官能化是最简单的反应之一,但它需要使用高活性和选择性的催化剂来完成这一反应过程。最近,使用结晶金属有机骨架(MOFs)和共价有机骨架(COFs)等多孔材料以及无定形多孔有机聚合物(POPs)作为C-H活性转化的新型非均相催化剂引起了广泛关注。这些多孔固体具有出色的结构均匀性、易调节性和永久性孔隙率。而且调整这些多孔材料的催化选择性可以通过设计其位点微环境来实现,例如金属节点替换、接头变化、节点/接头功能化和孔改性等,对于C-H官能化探究具有重要的研究意义。
02
成果背景
近日,权威期刊“CHEMICAL SOCIETY REVIEWS ”发表一篇题为“Metal–organic framework (MOF)-, covalentorganic framework (COF)-, and porous-organic polymers (POP)-catalyzed selective C–H bond activation and functionalization reactions”的综述,概述了 MOFs、COFs 和 POPs 作为各种 C-H 键活化反应的先进催化剂的最新技术,提供了有关它们的化学选择性、区域选择性和立体选择性控制的详细信息,并比较了它们的性能与其他催化剂一样,通过展示该领域目前的局限性和挑战来引发更多的研究,为未来的发展提供前景。
03
核心内容解读
金属有机框架 (MOF)
金属有机框架 (MOF),也称为多孔配位网络 (PCN) 或多孔配位聚合物 (PCP),是一种柔软的结晶多孔材料,近年来受到越来越多的关注。MOF 是多孔结晶、有机 -无机杂化材料,其中金属离子和有机多齿配体通过配位键连接在一起。由于其具有明确的孔隙结构、固有孔隙率、极大的表面积以及可调节的结构和功能,MOF 已被视为新兴的多相(光)催化剂。

图1. 通过金属离子/簇与有机接头/配体的自组装以及可能的 MOF 节点、接头和通过各种策略进行的孔改性来合成多孔 MOF 的示意图。 @ ROYAL SOCIETY OF CHEMISTRY
共价有机框架 (COF)
COF 被定义为扩展的多孔和结晶有机材料,通过动态共价化学形成具有不同性质的共价键。它们通常具有低密度、出色的耐用性以及高化学和热稳定性。它们的拓扑结构、结构、孔径和形状可以通过预先设计它们的组成单体轻松调整,从而具有良好的通用性。但是这些单体必须具有刚性骨架、合适的方向性,并且它们的缩合通常需要一定程度的可逆性才能获得高度有序的结构。
无定形多孔有机聚合物 (POP)
多孔有机聚合物 (POP)涵盖基于本综述中描述的缩合和偶联反应的所有有机材料家族。它们可以定义为一维、二维或三维的多孔材料,由具有预定拓扑和几何形状的有机构件组合而成,其通过共价键的连接赋予它们孔隙度。
多孔材料作为选择性C-H键活化反应催化剂的机理和行为
烷烃/C(sp3 )-H 脱氢/C-H 裂解
鉴于烯烃广泛用于合成不同的化学品,包括聚合物(例如聚苯乙烯、聚乙烯和聚丙烯)、化学中间体(例如乙苯和丙醛)和含氧化合物(例如丙酮和乙二醇)。烷烃的直接脱氢不仅需要极高的反应温度(即 500–700 ℃),副反应(即裂化和氢解)和焦化还会导致选择性低。 在温和的反应条件下,含有有机连接剂的多孔材料,包括 MOFs、COFs 和 POPs,成为潜在的催化剂或载体。

图2. (a) 催化剂活化形成 A-1 和 (b) 使用活化的模型物种 A-1 计算的 ODH (氧化脱氢)机理。 ΔH503K 单位为千卡每摩。@ ROYAL SOCIETY OF CHEMISTRY
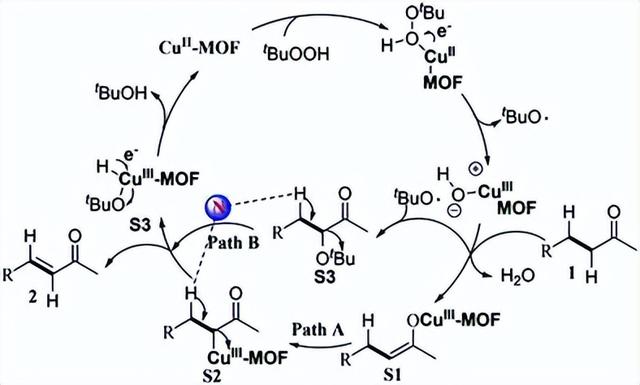
图3. Cu(ii)-MOF催化的饱和酮氧化α,β-脱氢反应路径。@ ROYAL SOCIETY OF CHEMISTRY
C(sp3 )–H/C(sp2 )–H 卤化
卤代有机化合物是制药工业和农业化学中重要的合成中间体和化学品。通过选择性 C-H 官能化进行卤化是合成卤代化合物的一种直接方法。
Cohen 的研究小组使用硫代儿茶酚酸螯合物固定的 Pd2 MOF 作为苯并[h]喹啉和 2-苯基吡啶选择性 C-H 键卤化的催化剂。硫醇官能化的 UiO-66 (UiO-66-TCAT) 是通过合成后接头交换合成的,其中 UiO-66 框架中 40% 至 70% 的对苯二甲酸酯接头被 2,3-二巯基对苯二甲酸 (tcatH2bdc) 取代。
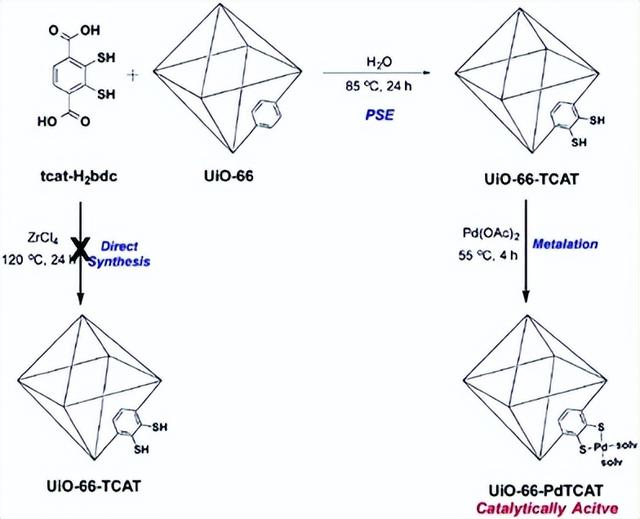
图4. UiO-66-TCAT 和 UiO-66-PdTCAT 的合成。@ ROYAL SOCIETY OF CHEMISTRY
相比之下,UiO-66-TCAT 的直接合成由于硫醇和羧酸酯基团对 Zr4 的竞争性螯合而失败。 配体交换后,Pd2 通过与固定的硫代儿茶酚 (UiO-66-PdTCAT) 螯合并入 UiO-66-TCAT 结构中,从而形成不饱和的单(硫代儿茶酚酸根)金属中心(图5)。掺入的 Pd2 -硫代儿茶酸酯复合物充当 C-H 官能化的活性位点。使用 UiO-66-PdTCAT 以 N-氯代琥珀酰亚胺 (NCS) 作为卤素源对苯并[h]喹啉进行卤化得到 95% 的产率,而在没有 Pd2 的情况下使用 UiO-66-TCAT 或 UiO-66 的对照实验表明相同条件下无活性。

图5. 光介导的异喹啉脱羧烷基化的机理建议。@ ROYAL SOCIETY OF CHEMISTRY
多种 C–X(X = C、O、N、B、S、P 和 Se)成键反应
碳-碳键形成反应
C(sp3)-C(sp3)偶联交叉,偶联反应是现代有机化学中的一类合成转化,为碳-碳(C-C)和碳-杂原子(C-X)的构建提供了可靠的平台。通过双惰性 C(X)-H 键活化键,不涉及卤代或拟卤代化合物。

图6. MIL-101(Cr)-SO3H 催化呫吨 (C(sp3)–H) 和亲核试剂 C(sp3)–H) 的需氧 CDC 反应。 @ ROYAL SOCIETY OF CHEMISTRY
C(sp3)-C(sp2) 偶联
除了激活两个 C(sp3)-H 物种进行碳-碳偶联反应外,其他类型的 C(sp3)-H 活化及其对 C(sp3)-C 的直接功能化 (sp2) 键的形成也有报道。

图7. 香豆素与 N,N-二甲基苯胺的交叉脱氢偶联,使用 VNU-20 MOF 形成 C(sp3)–C(sp2) 键。@ ROYAL SOCIETY OF CHEMISTRY
C(sp3)–C(sp) 偶联
炔丙基胺是有机化学中一类关键的化合物,具有广泛的药物和生物活性。炔丙基胺的直接合成方法之一是醛、胺和末端炔烃的三组分偶联(A3 偶联反应)。 当使用酮代替醛时,该反应称为 KA2 偶联。 炔丙胺可以通过仲胺或叔胺的 CDC 反应(C(sp3 )–C(sp) 偶联反应)获得,这也越来越多地被报道,与直接 C(sp3 )–H 键活化反应一致。其他合成炔丙胺的方法也有报道。这些方法涉及通过末端炔烃的 C-H 活化形成金属催化的 C-C 键。Phan 等人合成了多种 N-甲基-N- (3-苯基丙-2-炔基)苯胺通过MOF199催化的N,N-二甲基苯胺和末端炔烃之间的直接氧化C(sp3)-C(sp)偶联反应。MOF-199 是一种铜基 MOF, 使用这种 MOF 催化剂,N,N-二甲基苯胺通过激活 C(sp3)-H 键在二甲基乙酰胺 (DMA) 作为溶剂中成功进行了炔基化反应,在 120 ℃ 下 2.5 小时后产品收率达到 55-81%(图8)。 应当指出,该反应需要 TBHP 作为有机氧化剂。

图8. 采用 MOF-199 催化剂的 N,N-二甲基苯胺和苯乙炔之间的直接氧化 C(sp3)-C(sp) 偶联反应。@ ROYAL SOCIETY OF CHEMISTRY
C(sp2)–C(sp2) 偶联
通过直接激活两个 C(sp2)–H 键(芳烃的氧化偶联和芳烃的烯化/烯基化)的 C–C 偶联反应受到越来越多的关注。芳烃的氧化烯基化也称为 Fujiwara- Moritani 或氧化赫克反应。该反应的主要限制是 Pd(II) 催化剂的不稳定性和 C-H 键在不同位置的竞争性活化。因此,为了克服其中一些问题,一些研究小组开发了单中心多相催化剂 (SSHC)。2019 年,Farha 和同事将单离子 Pd(II) 位点锚定在两个节点上 通过溶液相溶剂热沉积 (SIM) 将合成后酸改性的 MOF 作为非均相载体,表示为 Pd@Hf-MOF-808-PO4 和 Pd@Hf-MOF-808SO4。Pd(II) 相互作用并稳定了 Hf6-oxo 节点上的酸功能化位点,显着防止它们聚集和失活为深棕色 Pd(0) 纳米颗粒 (NPs),突出了单中心多相催化剂 (SSHCs) 的作用。

图9. Pd(ii) 催化的氧化 Heck 偶联途径概述。@ ROYAL SOCIETY OF CHEMISTRY
C(sp)–C(sp) 偶联
氧化 Glaser–Hay 偶联反应是合成 1,3-二炔最有用的 C–H 键活化过程之一。该机理包括以下步骤:(i) 乙炔被碱去质子化,形成 Cu2 -乙炔化物; (ii) 空气中的 O2 将 Cu2 氧化成 Cu3 ; (iii) 将 Cu3 还原消除为 Cu 并形成产物,然后再生催化剂。
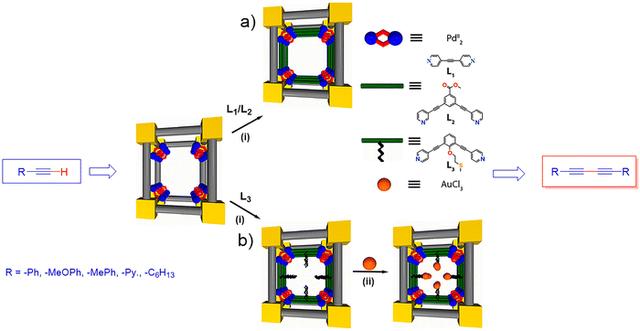
图10. SCCs@MOF 催化的炔烃的均偶联和交叉偶联(反应条件:催化剂、DABCO、EtOAc、室温和空气气氛下 14 小时)。它还展示了模板导向的方法,包括使用 PSM 合成 PdII8 和 PdII16 (a) 和异双金属 AuIII/PdII (b) 在初级 MOF 内机械键合的催化活性 SCC (SCCs@MOFs)。 (i) 配体 (L1-L3) 掺入和 (ii) 组装后金属化。@ ROYAL SOCIETY OF CHEMISTRY
碳-氮偶联
在过去的几十年中,金属介导的直接由 C-H 键形成 C-N 键的过程不仅已成为生成众多分子部分的有力方法,而且在许多有价值的合成中提供了广泛的应用。
α-酮酰胺作为有价值的结构基序是很有前景的分子,可用于医药和农产品的配方。基于 Cu(II) 之间的配位的蜂窝状 MOF离子和 2,5-二羟基对苯二甲酸 (H2dhtp),Cu-MOF-74,以 60% 的收率根据溶剂热法合成。随后,Cu 基 MOF 被用作高效的非均相苯乙二醛一水合物与不同胺在空气中在 80 ℃ 下脱氢交叉偶联反应 2 小时的催化剂(图11)。

图11. 在 Cu-MOF-74 存在下吡咯烷和苯基乙二醛的氧化偶联。@ ROYAL SOCIETY OF CHEMISTRY
碳-氧键形成反应
C-H 基团的氧化官能化是一个非常重要且用途广泛的反应,因为氧化产物是合成中间体并广泛用作进一步化学加工的构件。地球上丰富的天然气和气态碳氢化合物可以通过这个过程转化为易冷凝的燃料。然而,饱和的 C-H 键和芳族 C-H 具有热力学强度和动力学惰性,使得 C-H 的选择性氧化成为目前有机化学中最具挑战性的反应之一。
C(sp2/sp3)-H 硼基化
C-H 键的活化/硼基化在没有芳族化合物的情况下合成芳基硼酸及其酯由于其在有机化学中的广泛应用而引起了广泛关注。
碳-X 偶联(X = P、S 和 Se)
交叉偶联反应(CDC 反应)的概念也被开发用于 C-P 键的形成,可以使用 MOF 进行催化。

图12. FJI-Y2 MOF催化的碳-磷氧化偶联反应。@ ROYAL SOCIETY OF CHEMISTRY
涉及 C-H 键活化/功能化的串联/多步过程
受酶活性的启发,研究人员专注于开发人工级联(多米诺或串联)反应,其中多步转化在一锅中连续发生,而无需合成中间体的分离和纯化。 因此,串联反应符合绿色化学原则,减少了步骤数并最大限度地减少了后处理。这些通过 C-H 活化的连续多步反应通常需要具有两种或多种活性位点的多功能催化剂。

图13. Zn-PYI1 和 Zn-PYI2 MOFs,由 Zn SBUs 和 TPA 和 PYI 配体构成,用于苯丙醛的光催化不对称 α-烷基化。@ ROYAL SOCIETY OF CHEMISTRY
05
成果启示
该工作系统地介绍、分析和汇编了多孔无机和有机材料在 C-H 活化和 C-H 功能化领域的催化活性和选择性。这些材料(MOFs、COFs 和 POPs)表现出出色的各种结构和拓扑结构,表明了网状化学领域的成熟程度。文献中不仅报道了原始材料,而且还报道了功能化框架和复合系统,并在本综述中进行了汇总。这些策略中的每一个都有一些优点,但也有缺点。特别是,原始框架在构建模块的设计中需要额外的合成努力,但在可能的情况下,会产生均质且特征良好的材料。 相比之下,功能化由更简单的合成协议组成,但会导致材料的均质性降低。或者,复合材料在许多情况下会产生协同效应,但需要克服一些困难。
对本文提供的数据的分析表明,MOF 已被广泛研究,并且已经报道了许多催化系统。然而,有机材料的探索仍然较少,预计该领域将取得许多未来进展。
06
参考文献
Daliran S, Oveisi A R, Peng Y, et al. Metal-organic framework (MOF)-, covalent-organic framework (COF)-, and porous-organic polymers (POP)-catalyzed selective CH bond activation and functionalization reactions[J]. Chemical Society reviews.
https://doi.org/10.1039/D1CS00976A
,




