近日,复旦大学附属中山医院皮肤科杨骥主任团队发现了一种特殊的皮肌炎皮疹类型,被命名为假性血管性水肿。该研究成果近期已在线发表于皮肤科权威杂志——美国皮肤学会会刊(Journal of the American Academy of Dermatology,JAAD)上。这项研究成果以皮肤表型变化为线索对皮肌炎进行早期差异化诊疗,早期治疗可有效缓解重要内脏器官的损伤,减少致残和致死率,提高患者的生活质量和预后。
皮肌炎是一种可累及皮肤、肌肉和心肺等内脏器官的自身免疫性疾病。若累及肺可导致肺间质纤维化甚至呼吸衰竭,累及肌肉可导致肌痛肌无力、行动生活不能自理,吞咽困难等。虽然皮肌炎最早期的表现常常是皮肤的损伤,但它的皮肤表现形式复杂多样。因此识别皮肌炎的皮疹类型,对早期诊断、早期积极治疗、提高患者预后和生存率有非常重要的帮助。

假性血管性水肿的皮肌炎患者表现为面部、口唇、四肢为主的水肿,非凹陷性,伴或不伴红斑,皮疹无明显瘙痒。患者的皮疹看似临床常见的荨麻疹,但通过传统抗过敏治疗后疗效欠佳,容易被误诊、漏诊,进而导致治疗延误。若不及时控制病情,患者后续病情会进行性加重,部分患者会出现急进性肺间质纤维化,甚至发生重症肌炎,并累及吞咽肌群导致吞咽困难,预后较差。
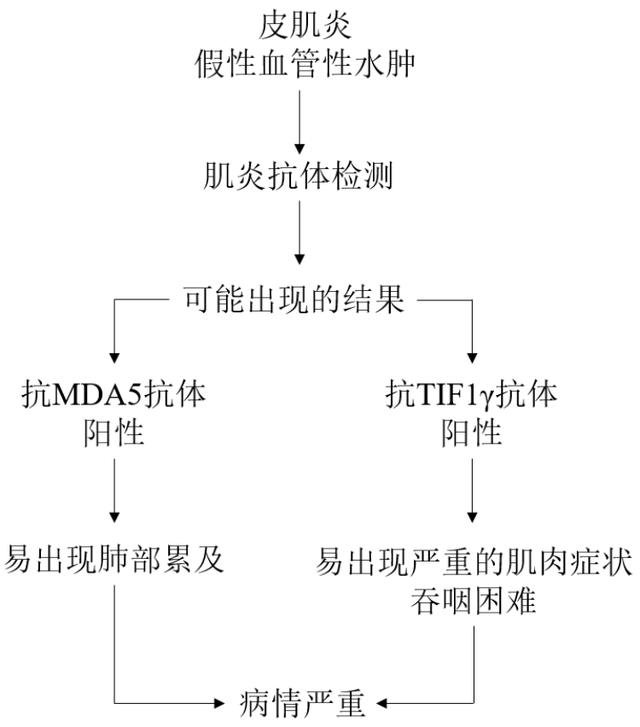
该研究通过对患者的深入分析,发现有假性血管性水肿的皮肌炎患者体内分别存在两种特殊的抗体,一种是抗黑色素瘤分化相关基因5抗体,另一种是抗转录中介因子1γ 。
患者如有假性血管性水肿及抗MDA5抗体呈阳性,几乎都会累及肺部,表现为肺间质纤维化,甚至会呈急进性肺间质纤维化,病情启动后预后较差。
患者如有假性血管性水肿和抗TIF1γ抗体呈阳性,则极有可能会出现严重的肌肉累及,表现为异常增高的肌酸激酶、肌痛和肌无力明显,严重影响患者生活质量,且此类患者容易发生吞咽困难。经治疗后,患者肌酶下降、肌力恢复,但吞咽困难的恢复较为棘手和迟缓,部分患者需借助胃管进食,后期容易发生继发感染。
当皮肌炎患者出现血管性水肿样皮疹时需要引起当事人和医生的高度重视,应进一步排查肌炎特异性抗体。皮疹结合抗体可对患者进行精准分型、评估及制定差异化个体化治疗方案,并预测患者的预后转归。
如有抗MDA5抗体阳性的患者需要密切关注肺部的损伤,如预判病情有进展可能,需尽早使用糖皮质激素、环磷酰胺等免疫抑制剂,和丙种球蛋白治疗;如抗TIF1γ抗体阳性的患者则需更加关注肌肉的损伤和吞咽困难的发生,可使用糖皮质激素、环孢素A等免疫抑制剂,和丙种球蛋白治疗。
此研究源于对临床患者的长期观察和随访,以及对大量的临床图片和资料收集、分析、归纳和总结,研究成果直接应用于指导临床诊疗,提高了对复杂疑难自身免疫性皮肤的诊疗水平。
下方示意图为皮肌炎合并假性血管性水肿患者的诊疗流程示意图:
论文通讯作者为杨骥主任医师,徐欣植、黄俊霞和王修远医师为共同第一作者,该研究得到了李明教授的指导和帮助。
来源:周到
,




