
多囊肾(Polycystic kidney disease,PKD)是一种遗传性肾脏疾病。 它会导致肾脏中形成充满液体的囊肿。 PKD可能损害肾功能并最终导致肾衰竭。 PKD是肾脏衰竭的第四大主要原因。 PKD患者也可能在肝脏中形成囊肿和其他并发症。
多囊肾病的发病原因是什么?PKD是遗传病,因此PKD的病因大多是遗传。 较少的PKD患者是由于患有其他严重肾脏问题的人中发展而来。 总的来说临床把PKD分成两种类型:
常染色体显性PKD常染色体显性遗传多囊肾病(ADPKD)有时也称为成人PKD。 根据芝加哥大学医学报道,ADPKD约占所有病例的90%,一般认为国内有150万左右患者。 在我国终末期肾衰竭病因中仅次于原发性肾小球肾炎、糖尿病肾病、高血压肾病之后,位于第四位。 常染色体显性遗传的特性意味着父母中若有一人患有ADPKD,则其后代将有50%的机会患上此病。
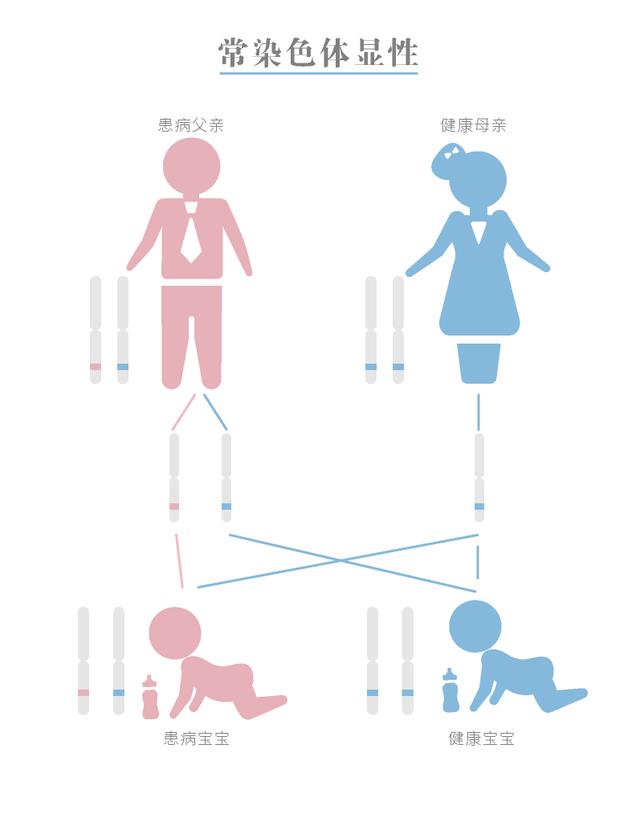
常染色体显性多囊肾病遗传方式
大多数成人型PKD患者的异常基因位于16号染色体的短臂,称为PKD1基因。 少数成人PKD患者的异常基因位于4号染色体的短臂,称为PKD2基因。 PKD1和PKD2基因分别编码完整的膜蛋白多囊蛋白-1 和多囊蛋白-2,两者结构相似,可发生相互作用。 PKD1或 PKD2 的突变可导致信号失调,环磷酸腺苷水平升高,最终导致囊肿生成。 成人型PKD症状通常会在30到40岁之后发生。 但是,有些人在儿童时期就开始出现症状。
常染色体隐性PKD常染色体隐性PKD(ARPKD)比ADPKD少见。 它也是遗传的,通常也称为儿童型PKD。致病基因是PKHD1,定位于6号染色体上,目前发现67种不同形式的突变。 但是父母双方都必须携带该疾病的突变基因才可能使后代患病,只携带一个PKHD1基因的人是不会得病的。
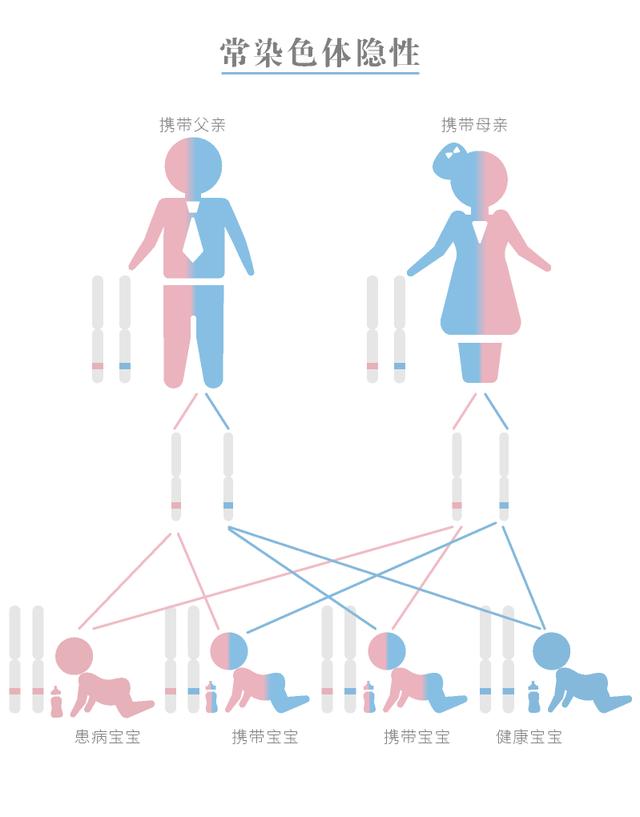
常染色体隐性多囊肾遗传方式
但是对于ARPKD的患者而言,一般在胎儿或者婴儿期肾功能就会出现下降,患者多在新生儿期死亡,存活的患者1年及10年的生存率分别为85%和82%。成活者25岁以前,大概70%的患者就进入到终末期肾衰竭。 因此对于常染色体隐形多囊肾病的患者,预后差,生存期短。
多囊肾病的症状是什么?许多人患有PKD多年,却没有出现与疾病相关的症状。 囊肿通常在人们开始注意到症状之前长出1.3cm或更大。 少数患者早期会出现腰腹疼痛、肉眼血尿或尿路感染等症状。表现特征可能因患者的合并症而异,并与其他疾病混淆。

心血管表现:高血压是多囊肾患者最常见的表现,并导致肾功能不全和左心室肥大等并发症,是多囊肾患者最常见的死亡原因。其他常见的心脏表现包括二尖瓣脱垂、继发于主动脉根部扩张的主动脉瓣关闭不全和心包积液

疼痛:疼痛是 ADPKD 患者最常见的症状,可表现为背部、胸部、腹部或腰部疼痛。 疼痛可能继发于囊肿破裂或肿大的肾脏压迫周围组织,但也可能是由于肾囊肿感染或肾结石所致。

尿路感染:症状性尿路感染在多囊肾患者中比一般人群中更常见,女性多见,病原体以革兰氏阴性菌为主。
肾囊肿感染时,尿培养以大肠埃希菌为主(74%-82.4%),其余为金黄色葡萄球菌、肠球菌、乳杆菌和厌氧菌等。
肾结石:多囊肾患者的肾结石的发生率(终生患病率约为 20%)是一般人群的 2 倍。 腰痛患者应怀疑肾结石。补液和止痛是一线治疗。必要时可采用体外冲击波碎石术和经皮肾镜取石术。

血尿:超过40%的多囊肾患者存在肉眼血尿,是肾功能不全快速进展的危险因素,尤其是发生在30岁之前时。
囊肿出血通常在一周内消退,无需治疗,尽管仍可能存在镜下血尿。

腹部表现:肾外囊肿可累及精囊、卵巢、胰腺、脾脏和中枢神经系统,但以肝囊肿最常见。患者通常无症状,且肝功能正常(除血清碱性磷酸酶轻度升高)。由于雌激素促进肝囊肿的增殖,在妊娠和雌激素治疗期间,肝囊肿可能迅速增大。多囊肾的其他腹部表现包括憩室病、腹疝和腹股沟疝。

颅内动脉瘤:颅内动脉瘤是多囊肾患者最严重的肾外表现。多囊肾患者发生颅内动脉瘤的可能性至少是正常人群的 2 倍;有颅内动脉瘤家族史的多囊肾患者发生率更高。
颅内动脉瘤的并发症包括血栓栓塞、占位效应的局部颅内压升高和致命性蛛网膜下腔出血。直径小于 7 mm 且在前循环中的动脉瘤破裂可能性小,并且可以监测。建议在有指征时使用时间飞跃法磁共振血管造影行动脉瘤筛查。
多囊肾病的诊断影像学诊断尽管 CT/MRI 检测肾囊肿的灵敏度略高,但 B 超成本较低、不需使用造影剂和无辐射暴露,是首选的筛查方式。
CT 和 MRI 在筛查超声无法诊断的病态肥胖患者中发挥作用。ADPKD 的 B 超诊断标准列于表 1。
|
年龄(岁) |
肾脏囊肿数量 |
|
存在 1 型 ADPKD 风险者(基于 Ravine D et al, 1994) | |
|
<30 |
单侧/双侧肾 ≥ 2 个 |
|
30-59 |
单侧肾 ≥ 2 个 |
|
≥ 60 |
单侧肾 ≥ 4 个 |
|
存在风险,未知基因型者(基于 Pei Y et al, 2009) | |
|
15-39 |
单侧/双侧肾 ≥ 3 个 |
|
40-59 |
单侧肾 ≥ 2 个 |
|
≥ 60 |
单侧肾 ≥ 4 个 |
表 1 ADPKD 的影像学诊断标准(B 超)
分子诊断对于缺乏家族史或影像学检查无法确诊的多囊性肾病 (PKD) 患者,分子诊断意义重大,可通过分析患者是否存在 PKD1 及 PKD2 基因突变而明确诊断。
主要的技术有基因连锁分析、直接检测基因突变、单链构象多态性分析以及变性高效液相色谱 (DHPLC) 等,其中 DHPLC 是前几年较成熟且应用较普遍的基因诊断方法。
多囊肾病治疗方法一览监测ADPKD进展主要指标是总肾体积及肾小球滤过率。 随着基因诊断及植入前诊断(PGD)技术进步,给阻断ADPKD遗传带来了广阔的前景。 ADPKD治疗以对症为主,特殊治疗药物正在深入研究中。
阻断遗传长期以来,ADPKD主要依靠药物治疗控制疾病进展,但近年来三代试管婴儿技术的不断发展,使我们可以通过植入前基因诊断技术从根本上阻断ADPKD的遗传。

KDIGO指南推荐使用支持治疗以减轻ADPKD临床症状,减少并发症发生率及病死率。下面我们重点介绍两方面常见的支持治疗:
高血压治疗高血压通常在慢性肾脏病出现临床症状前发生。良好的控制血压是PKD患者治疗的关键部分。eGFR>60mlmin·(1.73m2)-1,的18-50对患者,血压控制目标值< 110/75 mmHg,其他患者血压控制目标值:< 130/80 mmHg。

囊肿感染可出现腰痛,高热和/或感染性休克。肾囊肿感染难以有效治疗,因抗生素无法渗入较大囊肿。 脂溶性抗生素,如喹诺酮类、服方新诺明及甲硝唑等是首选治疗药,因相比青霉素,该类药物的扩散速度更快。治疗至少持续10-14天,或至症状消失、体温正常、两次血/尿培养结果阴性后1周停药。
抑制囊肿生长近年来,临床研究主要集中于特异性抑制囊肿生长的药物,包括血管加压素2受体拮抗剂、mTOR抑制剂、生长抑素类似物等。
目前唯一批准ADPKD患者临床使用的是血管加压素2受体拮抗剂托伐普坦,可有效阻断细胞内源性cAMP通路以抑制囊肿生长和囊液分泌。该药已在欧盟、英国、日本、加拿大和韩国获批使用,主要用于控制高风险ADPKD患者的肾病进展。


PKD患者自进展为慢性肾脏病开始就应开始随访治疗。 终末期肾病患者可进行肾脏替代治疗(透析和移植)。 若多发性肾脏囊肿较大可进行手术切除单侧或双侧肾脏,继而进行肾移植。活体肾捐赠者应进行全面筛选,确保捐赠者无PKD。
日常生活需要做好哪些预防工作?预防外伤多囊肾囊肿的不断肿大,将会导致囊肿的囊内压不断增高,迫使患者的双肾也不断增大,腹腔内压加大。如此时任何一点轻微的外伤,如扭伤,碰伤,跌伤等就会加大腹脏内压或外伤外力直接对肿大囊肿的冲击,促使具有高内压的囊肿破裂、出血,很易诱发感染。
预防感冒患有多囊肾疾病的病人内心是非常痛苦的,因为同别的肾病不一样,多囊肾是一种终身性的遗传疾病,需一辈子伴随,既便是格外注意、家人的体贴照顾再多,仍阻挡不了囊肿继续肿大的客观现实。此时,如患感冒,尤其是反复感冒就会使得多囊肾病人的肾损害加重一分,起到雪上加霜的恶化作用,更会加速肾功能损伤的进展。
控制好高血压多囊肾患者在肾功能受损之前就会出现高血压,高血压的出现会加速肾功能的损害,同时高血压也会对心、脑血管的损伤,会对多囊肾伴有脑血管瘤破裂出血造成中风等的严重并发症,故控制好血压对延缓肾功能恶化速度,防止并发症至关重要。
控制好饮食多囊肾患者的合理治疗饮食对控制肾功能恶化进展非常重要。采用低盐饮食,每天2~3克食用盐为宜,少吃含钾、磷高的饮食,要低蛋白、低脂肪饮食,多吃富含维生素与植物粗纤维饮食,保持大便通畅。
温馨提示,以下食品应避免食用:
1.发酵性食品:这里所讲的发酵食品主要是菌变发酵的食品,如豆腐乳、臭鸡蛋类。食用此类食物会加剧囊肿生长速度。所以,预防多囊肾,不要吃发酵性食品。
2.高蛋白食品:预防多囊肾的饮食中,都应该低蛋白饮食,避免体内氮类代谢物合成,减轻肾脏的排泄力,如大豆、豆腐、以及其他豆类制品。
3.酒类饮料:酒类、特别是白酒,对于多囊肾患者来讲宜戒掉,酒类对与肾脏的刺激性优为重要。它可以刺激多囊蛋白活性,加速囊肿生长,因此预防多囊肾时,饮食中尽量减少酒类饮料。
4.内脏食品:动物的内脏作成的熟食千万不要食用,特别是动物肝脏,在宰杀动物过程中很多毒素遗留在肝、肾内,特别是肝脏,肝脏的功能就是解毒,很多动物代谢的毒素都遗留在内脏中,如果病人服用这些物质后无形对肾脏增加负担,加重病情。
,




