Hi,大家好,我是晨曦
不知道大家有没有这样的烦恼,就是感觉有很多东西没有学到,但是每当学的时候又会感觉无处可学,那么这个时候可能提醒你需要进一步“鞭策”自己了
本期开启一个全新的栏目——文章复现内容,其实也不是传统意义上的全文复现了,晨曦找了一些有意思且质量较高的文献,这些文献往往提供了配套的代码,所以允许我们可以下载下来进行学习,毕竟观看大佬写的代码是一种快速进步的方式
那么,我们就来看一下今天这期我们想要学习的Cell文章,如下:

文章的具体内容我们就不过多探讨了,当然不愧是Cell,作者很贴心的在后面的method部分放上了自己的代码以及数据链接,重点是作者整理的代码逻辑非常的清晰!!(强烈好评)

那么接下来先做一个简单的预告,我们通过这篇推文可以学到什么QAQ
第一:你可以学会下面的这组可视化结果

第二:你可以学到在Rstudio中使用python的一些简单技巧
第三:你可以学到一些有意思的单细胞处理技巧
第四:你可以学到基于随机森林计算评分的技巧
除此之外,我还为大家整理了许多单细胞测序的高分套路模板和文章复现资源,想要的小伙伴可以点击了解详情免费领取哦~
滴滴,如果你感兴趣上面四点,那么我们就开始吧
#准备工作library(MAST)# BiocManager::install("dtm2451/dittoSeq@a3bfe2b")library(dittoSeq)library(caret)library(ranger)# devtools::install_github("vqv/ggbiplot")# devtools::install_github("enriquea/feser")library(feseR)library(pROC)# install.packages("reticulate")library(reticulate)# use_python("/anaconda3/envs/r-reticulate/bin/python")# # Run this in Terminaluse_condaenv("seurat", required = TRUE)# # conda create -y -c conda-forge -n r-reticulate umap-learn=0.3.10# devtools::install_version(package = "adehabitat", version = "1.8.20")# devtools::install_version(package = "SDMTools", version = "1.1-221.1")# devtools::install_version(package = "Seurat", version = "3.0.2")library(Seurat)
作者很贴心的把需要下载的R包命令进行了注释,同时因为我们后期可能会在Rstudio环境中使用Python,所以作者也把配置Python环境的相关操作进行了注释
所以这里,我们学到了一点:Rstudio中通过使用reticulate包可以调用Python环境,当然Rstudio进行更新后其实也是可以创建python脚本了(Ps:但是还是习惯Python使用Jupyternotebook)
#读取输入数据(标准的10×下机数据)Tcells <- CreateSeuratObject(Read10X("文献复现/GSE158492数据集/10×/"))#物种人
样本信息简单的介绍一下:
物种:人
细胞来源:来自10个独立样本的CD4、CD8、CD34 造血干细胞和祖细胞
技术:10×genomic
Tcells[["percent.mito"]] <- PercentageFeatureSet(Tcells, pattern = "^MT-")#计算线粒体基因Tcells[["percent.ribo"]] <- PercentageFeatureSet(Tcells, pattern = "^RPS|^RPL")#计算红细胞
这里作者选择的质控标准如下:
with at least 750 genes
with at least 1500 UMIs
with less than 5% mitochondrial UMIs
可以很清楚的看到,在线粒体基因的角度,质控标准是比较严格的
Tcells.cut <- subset(Tcells, subset = nFeature_RNA > 750)Tcells.cut <- subset(Tcells.cut, subset = nCount_RNA > 1500)Tcells.cut <- subset(Tcells.cut, subset = percent.mito < 5)
下面作者进行了双细胞的去除,但是这里晨曦说一下个人的观点,因为我们平时也会接触到scRNA-seq的分析,但是我们很少会进行双细胞的去除,或者说这一步骤如果不进行其实对我们的结果应该也没有太大的改变因为我们的质控标准是选不完的,我们晨曦认为:我们在进行自己的单细胞测序的时候,只需要对线粒体和红细胞以及细胞周期进行质控即可,个人理解,仅供参考
#去除双细胞Tcells.cut <- importDemux( Tcells.cut, demuxlet.best = c("文献复现/GSE158492数据集/GSE158492_CD4.best.txt", "文献复现/GSE158492数据集/GSE158492_CD4-8.best.txt", "文献复现/GSE158492数据集/GSE158492_CD8.best.txt"), lane.names = c("CD4","CD4-8","CD8"))Tcells.cut[["Sample"]] <- sapply( meta("Sample",Tcells.cut), function(X) strsplit(X, split = "CD4_")[[1]][2])Tcells <- subset(Tcells.cut, subset = demux.doublet.call == "SNG")
这里可能会有小伙伴疑问:importDemux函数有什么作用?这个函数其实就是把多个管道的样本信息导入到我们的S4对象中,然后可以通过解卷积的方法帮助我们识别双细胞状态的油包水样结构参考教程:Instructional tutorial for using demuxlet (protocols.io)
#然后下面作者又进行了一些针对metadata的操作#这一部分的操作在我们后续的分析中,我们不是必须要添加Tcells@meta.data$age <- "unknown"Tcells@meta.data$age[grep("FS",Tcells$Sample)] <- "fetal"Tcells@meta.data$age[grep("UCB",Tcells$Sample)] <- "cord"Tcells@meta.data$age[grep("APB",Tcells$Sample)] <- "adult"# Add Tcelltype# This is dependent on both sample-assignment and 10X-lane, so I will need to add how samples were added to each lanesamples.4 <- c("FS3", "FS4", "FS5", "UCB1", "UCB2", "UCB5", "APB1", "APB2", "APB4", "APB5")samples.8 <- c("FS1", "FS5", "UCB2", "UCB3", "UCB4", "UCB5", "APB2", "APB3", "APB5")samples.48.4 <- c("FS1", "FS2", "UCB4", "UCB3", "APB3") samples.48.8 <- c("FS3", "UCB1", "APB1", "APB4")samples.48 <- c(samples.48.4,samples.48.8)Tcells@meta.data$Tcelltype <- NATcells@meta.data$Tcelltype[(Tcells$Sample%in%samples.4) & (Tcells$Lane=="CD4")] <- "CD4"Tcells@meta.data$Tcelltype[(Tcells$Sample%in%samples.48.4) & (Tcells$Lane=="CD4-8")] <- "CD4"Tcells@meta.data$Tcelltype[(Tcells$Sample%in%samples.8) & (Tcells$Lane=="CD8")] <- "CD8"Tcells@meta.data$Tcelltype[(Tcells$Sample%in%samples.48.8) & (Tcells$Lane=="CD4-8")] <- "CD8"Tcells@meta.data$Tage <- NATcells@meta.data$Tage[Tcells$Tcelltype=="CD4"&!is.na(Tcells$Tcelltype)] <- paste0("4-",Tcells$age[Tcells$Tcelltype=="CD4"&!is.na(Tcells$Tcelltype)])Tcells@meta.data$Tage[Tcells$Tcelltype=="CD8"&!is.na(Tcells$Tcelltype)] <- paste0("8-",Tcells$age[Tcells$Tcelltype=="CD8"&!is.na(Tcells$Tcelltype)])sum(is.na(Tcells$Tage))Tcells@meta.data$Tage[is.na(Tcells$Tage)] <- "0"sum(Tcells$Tage=="0")Tcells <- subset(Tcells, subset = Tage != "0")
进行完上述的步骤,我们可以保证每一个细胞ID都有一个标签,也就是说上面的内容其实就是进行了数据质控的部分
#细胞周期s.genes <- cc.genes$s.genesg2m.genes <- cc.genes$g2m.genesTcells <- CellCycleScoring(Tcells, s.features = s.genes, g2m.features = g2m.genes, set.ident = TRUE)#细胞周期信息储存在CellCycleTcells@meta.data$CellCycle <- Idents(Tcells)Idents(Tcells) <- "Lane" #idents函数可以直接支持赋值metadata内的变量信息,然后就可以调用该变量内的信息内容#idents函数展示的结果其实就是Barcode ID和某lab的对应关系,如果进行完细胞注释,默认就是与细胞注释的对应关系
然后下面我们就可以进行数据标准化、寻找高可变、数据归一化、PCA等标准常规操作,我们这里就加快的步伐~
Tcells <- NormalizeData( object = Tcells, normalization.method = "LogNormalize", scale.factor = 10000, verbose = F)Tcells <- FindVariableFeatures(object = Tcells, verbose = F, nfeatures = 2000)Tcells <- ScaleData( object = Tcells, vars.to.regress = c("CellCycle", "percent.mito", "nCount_RNA"), verbose = F)Tcells <- RunPCA( object = Tcells, verbose = T, npcs = 50)Tcells <- RunTSNE( object = Tcells, reduction.use = "pca", dims = 1:9, seed.use = 1)Tcells <- FindNeighbors(Tcells, reduction = "pca", k.param = 20, dims = 1:9)Tcells <- FindClusters(Tcells, modularity.fxn = 1, algorithm = 1, resolution = 0.1)Tcells <- RunUMAP( object = Tcells, reduction = "pca", dims = 1:9, seed.use = 1)
然后我们进行可视化的部分
dittoDimPlot(Tcells, "Tage", size = 1, reduction.use = "umap", colors = c(1:3,9:11), main = "T cells Lineage and Stage", rename.var.groups = c("Adult-CD4", "Fetal-CD4", "UCB-CD4", "Adult-CD8", "Fetal-CD8", "UCB-CD8"))
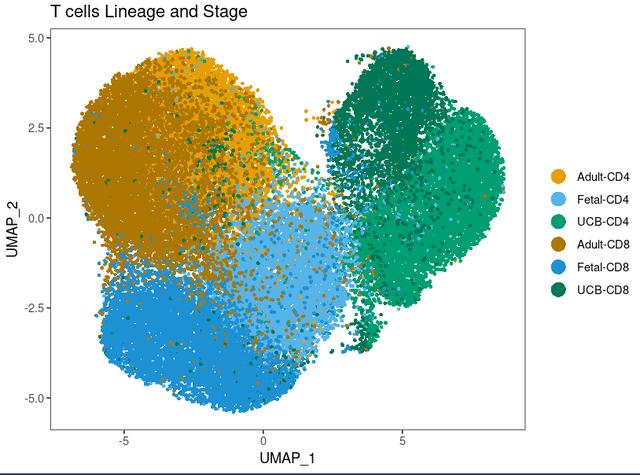
可以很清楚的看到我们其实是把不同的label绘制在了一张可视化上,然后我们想要像Cell上在周边展示不同label分别的可视化,在R中我们可以这么运行代码
#创建一个list列表,把所有的可视化装进去plots <- list( dittoDimPlot(Tcells, "Tage", size = 0.5, reduction.use = "umap", cells.use = Tcells$Tage=="4-adult", colors = 1, legend.show = FALSE, ylab = NULL, xlab = NULL, main = NULL, show.axes.numbers = FALSE), dittoDimPlot(Tcells, "Tage", size = 0.5, reduction.use = "umap", cells.use = Tcells$Tage=="4-cord", colors = 2, legend.show = FALSE, ylab = NULL, xlab = NULL, main = NULL, show.axes.numbers = FALSE), dittoDimPlot(Tcells, "Tage", size = 0.5, reduction.use = "umap", cells.use = Tcells$Tage=="4-fetal", colors = 3, legend.show = FALSE, ylab = NULL, xlab = NULL, main = NULL, show.axes.numbers = FALSE), dittoDimPlot(Tcells, "Tage", size = 0.5, reduction.use = "umap", cells.use = Tcells$Tage=="8-adult", colors = 9, legend.show = FALSE, ylab = NULL, xlab = NULL, main = NULL, show.axes.numbers = FALSE), dittoDimPlot(Tcells, "Tage", size = 0.5, reduction.use = "umap", cells.use = Tcells$Tage=="8-cord", colors = 10, legend.show = FALSE, ylab = NULL, xlab = NULL, main = NULL, show.axes.numbers = FALSE), dittoDimPlot(Tcells, "Tage", size = 0.5, reduction.use = "umap", cells.use = Tcells$Tage=="8-fetal", colors = 11, legend.show = FALSE, ylab = NULL, xlab = NULL, main = NULL, show.axes.numbers = FALSE), dittoDimPlot(Tcells, "Tage", size = 1, reduction.use = "umap", legend.show = FALSE, color.panel = dittoColors()[c(1:3,9:11)], main = NULL), dittoSeq:::.grab_legend(dittoDimPlot(Tcells, "Tage", size = 1, reduction.use = "umap", color.panel = dittoColors()[c(1:3,9:11)], rename.var.groups = c("Adult-CD4", "UCB-CD4", "Fetal-CD4", "Adult-CD8", "UCB-CD8", "Fetal-CD8"))))pdf("Tcells-Figs/Tcell_umap_surround.pdf", w=6, h=6)gridExtra::grid.arrange(grobs = plots, layout_matrix = matrix(c( 7,7,7,4, 7,7,7,5, 7,7,7,6, 1,2,3,8), ncol = 4))dev.off()
dittoDimPlot函数绘制细胞聚类图中里面有一个参数是cells.use可以选择细胞ID进行可视化grid.arrange函数可以帮助我们进行排版,这里重点解释一下矩阵对于图形摆放的问题我们都知道如果layout_matrix这么写:matrix(c(1,1,2,3),2,2,代表了绘制一个两行两列,一共三张图,第一张图占据一行,后面两张图加起来占据一行那么如果是这种矩阵的形式其实也是很好理解的,就是一个占位的问题
layout_matrix = matrix(c( 7,7,7,4, 7,7,7,5, 7,7,7,6, 1,2,3,8), ncol = 4))#我们list里面一共有8个plot,那么我们将用很大一部分去绘制第七张图,然后右边从上到下分别绘制第4、5、6、8张图,然后下面从左到右绘制1、2、3图,所以就是下面这个样子

至此,我们成功完成了Figure3-A的复现,那么我们接下来继续
#绘制样本细胞百分率dittoBarPlot(Tcells, "ident", group.by = "Sample", x.reorder = c(6:8,9:13,1:5), main = NULL, cells.use = Tcells$Tcelltype=="CD8", ylab = "Fraction of CD8\nin each cluster", legend.show = FALSE, legend.title = "Clusters", x.labels = c(paste0("F",1:5),paste0("U",1:5),paste0("A",1:5))[c(1,3,5:15)], x.labels.rotate = T, xlab = NULL)

那么到这里主要的两张可视化技巧我们就学到这里,我们看文献的时候,还比较感兴趣的就是通过机器学习算法构建评分,也就是方法学中下面这部分的内容

那么通过阅读方法学部分,我们可以了解到大概分以下几个步骤:
with at least 750 genes
with at least 1500 UMIs
with less than 5% mitochondrial UMIs
1. Pick out a 10% of fetal and adult cells training set(选出10%细胞数量作为训练集)
2. Calculate the FvA markers for that set(计算差异基因)
3. Run correlation and random-forest feseR to narrow down the genelist.(使用机器学习算法缩小预测变量范围)
4. Generate RFmodels based on feseR-restricted genesets(产生随机森林模型)
5. Check accuracy in fetal vs adult cells that were not in the training set(检验模型的性能,并不是在训练集上,而是测试集)
Score UCB (refered to as "cord" within the object)(因为这个模型构建的时候我们选择的是两种状态的细胞,然后我们进行测试评分的时候其实会有第三种细胞,这个时候我们可视化可以得到一种连续的状态,说明了第三种细胞很有可能就是第一种细胞和第二种细胞的中间过渡态)
set.seed(1909)Idents(Tcells) <- "age"#fetal-cluster UCB-cluster adult-cluster # 14542 10921 14689 inTraining <- createDataPartition(Idents(Tcells), p=0.1, list = FALSE)#选择10%的细胞作为训练集inTraining <- inTraining[Idents(Tcells)[inTraining]%in%c("fetal","adult")]#构建训练集的时候我们只需要两种状态的细胞age.inTrain <- Tcells$age#提取细胞ID和label的对应信息age.inTrain[-inTraining] <- 0 #所有没有被我们选中的把label清空Idents(Tcells) <- age.inTrain#训练集作用载数据集上#这个时候我们的数据集应该是包含了一部分有label和很大一部分没有label#寻找差异基因并进行筛选FvA <- FindMarkers(Tcells, ident.1 = "fetal", ident.2 = "adult", test.use = "MAST")FvA_padjFC <- FvA[abs(FvA$avg_log2FC)>=0.585 & FvA$p_val_adj<0.05 & !(is.na(FvA$p_val_adj)),]markers <- rownames(FvA_padjFC)#获取我们真正的训练集合training <- as.matrix(t(GetAssayData(Tcells)[markers,inTraining]))
这里我们需要注意GetAssayData函数的书写方式,以前我们都是直接添加对象就可以提取S4对象内的表达矩阵或者说是我们最常提取的counts矩阵,但是我们也可以提供一个观测为基因,变量为细胞ID或者数值,然后我们能做到就是提取观测和变量范围内的表达矩阵
training[1:10,1:5]# JUNB RPS24 ZFP36L2 TMSB4X DUSP1#AAACCTGAGATAGGAG-1 2.191085 4.319520 1.840011 4.354152 1.294263#AAACCTGCAAGCGTAG-1 1.707412 4.262362 0.000000 4.754660 2.050529#AAAGATGAGAGGGCTT-1 4.418952 3.535619 3.458002 4.664140 2.420471#AAAGATGCAAGCTGTT-1 4.481459 3.564488 3.256595 4.149503 2.601244#AAAGATGCATGTCCTC-1 4.064382 3.222724 2.776347 3.614818 1.949347#AAAGATGCATTCACTT-1 4.153450 3.432844 2.999145 4.195355 2.682409#AAAGATGTCATCGGAT-1 4.610814 3.064502 3.531894 4.660339 3.064502#AAAGCAAAGAAACGAG-1 3.924906 4.003429 2.227292 3.251311 2.227292#AAAGCAACAGATCCAT-1 2.850958 4.107304 1.313022 5.066440 1.313022#AAAGCAACATGTCCTC-1 1.286435 3.759285 0.000000 5.258795 1.286435#创建结局变量Train_val <- array(1, length(inTraining))Train_val[Tcells$age[inTraining]=="fetal"] <- 0#去除具有相关性的预测变量training.trim <- filter.corr(scale(training), Train_val, mincorr = 0.3)#通过递归随机森林筛选预测变量feser <- rfeRF( features = training.trim, class = Train_val, number.cv = 3, group.sizes = seq_len(ncol(training.trim)), metric = "ROC", verbose = FALSE)#文献中使用的10折,但是运行速度会很慢,即使是3折也需要大概20min左右的时间markers.feser <- feser$optVariableslength(markers.feser)#[1] 84
我们的训练集一共有104个预测变量,通过算法我们得到了84个预测变量,但是实际上我们并不可能把这84个全部纳入到模型之中,所以我们需要结合生物学背景或者其它算法来帮助我们进一步筛选
feser$results#查看结果

这个时候我们可以通过结果理解了算法本身的过程,这个算法就是一开始是全部预测变量然后不断消除预测变量的数量,然后计算出来ROC以及sensitivity和specificity等信息,这个时候按照作者的理解,作者发现在第17个预测变量纳入的时候可以保证ROC、sensitivity和specificity均超过0.99,所以作者选择了17个预测变量作为后续的分析内容自动特征选择用于构建不同子集的许多模型,并筛选出有用特征从算法上来看,应该是三折每一次都选择不同的特征子集只不过是数量是一致的,但是如果选择特征子集是具有算法的,所以说基本上选择并不会出现太大的偏差
vars17 <- unique(feser$variables$var[feser$variables$Variables==17])#提取选择的17个预测变量#[1] "RPS24" "JUNB" "TMSB4X" "ZFP36L2" "TXNIP" "HSP90AA1"#[7] "MT-CO1" "MYL6" "DUSP1" "GAPDH" "PFN1" "SOX4" #[13] "CD52" "TSC22D3" "YBX1" "KLF6" "ACTB" "CRIP1" #[19] "RGS1"
这里之所以是19个预测变量就是晨曦前面说过,整体组合基本是固定的,因为固定数量下最可以代表数据集的形式是特定的,但是仍然会存在一些细微的偏差,这个随着预测变量数量越多越明显,就好像一个群体,你要选择最优秀的人,那么这个人其实很好选,但是如果你要选择最优秀的十个人,那么很显然就会有比一个人更多的可能
(vars17.counts <- sapply(vars17, function(X) length(grep(X, feser$variables$var[feser$variables$Variables==17]))))#获取每个预测变量被纳入的次数#因为我们是三折,所以如果在三个训练集中均被纳入那么很显然这个预测变量会更好

可以很清楚的看到,SOX4被纳入两次,那么很有可能第三次的时候纳入的就是CD52,CRIP1被纳入2次,那么第三次很有可能就是RGS1,那么这四个预测变量可以划归为两个,然后数量正好就是17,所以这个算法的运算逻辑就是这样
#根据迭代次数选择最合适的预测变量(vars.use <- names(head(vars17.counts[order(vars17.counts, decreasing = TRUE)], 17)))#[1] "RPS24" "JUNB" "TMSB4X" "ZFP36L2" "TXNIP" "HSP90AA1"#[7] "MT-CO1" "MYL6" "DUSP1" "GAPDH" "PFN1" "TSC22D3" #[13] "YBX1" "KLF6" "ACTB" "SOX4" "CRIP1"
那么,到这里预测变量就选择完毕了,然后我们继续
markers.feser <- vars.usetraining <- as.matrix(t(GetAssayData(Tcells)[markers.feser,inTraining]))#训练集进行重整#创建终点结局变量Train_val <- array(1, length(inTraining))Train_val[Tcells$age[inTraining]=="fetal"] <- 0#拟合模型rf_mod <- train(Train_val ~ ., set.seed(998), data= cbind(training,Train_val), method = "ranger", metric = "MAE", trControl = trainControl(method = "cv", number = 3, repeats = 3), tuneGrid = expand.grid(mtry = round(length(markers.feser)*.75,0), splitrule = c("extratrees"), min.node.size = 1) )#获得每一个细胞的打分并存储在metadata中Tcells@meta.data$RFScore <- as.double(predict(rf_mod,t(GetAssayData(Tcells)[markers.feser,])))#标记终点结局标签Tcells@meta.data$inTraining <- FALSETcells@meta.data$inTraining[inTraining] <- TRUE #我们选择的训练集则是TRUE#验证打分准确性Idents(Tcells) <- "age"roc_obj <- roc(response = as.numeric(meta("ident",Tcells)[!(Tcells$inTraining) & meta("ident",Tcells)%in%c("fetal", "adult")]=="adult"), predictor = Tcells$RFScore[!(Tcells$inTraining) & meta("ident",Tcells)%in%c("fetal", "adult")], plot = T)auc(roc_obj)#Area under the curve: 0.9998
这块我们学习到的技巧就是可以把细胞亚群的label作为我们的终点结局进而来构建模型,之所以构建随机森林则是因为其对数据最为宽容
#可视化打分dittoPlot(Tcells, "RFScore", cells.use = Tcells$inTraining, group.by = "Sample", color.by = "age", plots = c("jitter","vlnplot"), boxplot.color = "white", boxplot.fill = F, vlnplot.lineweight = 0.3, vlnplot.width = 3, sub = "in Training", colors = c(1,3))### 7. Check the look for all T cells.dittoPlot(Tcells, "RFScore", cells.use = !(Tcells$inTraining), group.by = "Sample", color.by = "age", plots = c("jitter","vlnplot"), boxplot.color = "white", boxplot.fill = F, vlnplot.lineweight = 0.3, vlnplot.width = 5, sub = "NOT in training")dittoDimPlot(Tcells, "RFScore", cells.use = !(Tcells$inTraining), sub = "NOT in training", size = 2, reduction.use = "umap")
这块图片就不展示了,因为和文献中是一样的,这块的技巧告诉我们,只需要把我们想要的信息添加到metadata中,那么后续的可视化就可以直接调用那么至此,这篇文献的一些关于单细胞可视化以及机器学习算法还有一些分析细节到这里就结束了,感觉收获满满,大家也快找到数据快速练习吧那么本期推文到这里就结束了,感兴趣这个专题的小伙伴也欢迎大家在评论区留言,然后以后会多多更新相似专题哦
别忘了点击了解详情领取我为大家整理了许多单细胞测序的高分套路模板和文章复现资源,全部免费!
1.GitHub - dtm2451/ProgressiveHematopoiesis: Code to go along with a manuscript: Bunis, et. al., Cell Reports, 2021.
2.Bunis, D. G., Bronevetsky, Y., Krow-Lucal, E., Bhakta, N. R., Kim, C. C., Nerella, S., Jones, N., Mendoza, V. F., Bryson, Y. J., Gern, J. E., Rutishauser, R. L., Ye, C. J., Sirota, M., McCune, J. M., & Burt, T. D. (2021). Single-Cell Mapping of Progressive Fetal-to-Adult Transition in Human Naive T Cells. Cell Reports, 34(1). https://doi.org/10.1016/j.celrep.2020.108573
,