病例1
患儿,男,1岁7个月,因智力运动发育落后就诊。母孕期无异常,患儿系第1胎第1产,足月剖宫产,出生体重3.9kg,生后哭声弱,阿普加评分不详,生后反应差诊断为“缺氧缺血性脑病”住院治疗7天。无病理性黄疸史。生后喂养困难,体重增长慢,无抽搐,无运动倒退病史。主动表达少,仅会发单音词,注意力不集中,注视时间短;不会独走,独坐稳,会爬,可扶站。精细动作差,可拇食指对捏,动作笨拙;对视、追视尚可,追听欠灵敏,可逗笑出声,认生人。
查体:
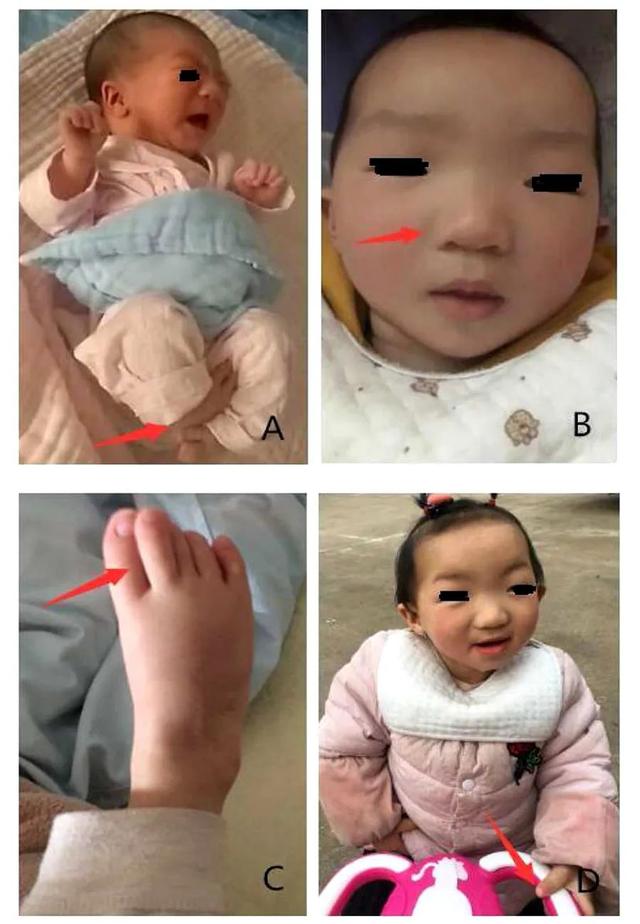
身长85cm,体重10kg,头围47.2cm,神情呆滞,长颅、窄脸、睑裂狭小、球形鼻尖、上唇薄、小下颌、腭弓高(图A、B),拇趾长,第3足趾痉挛(图C、D)。心肺腹未见异常,双侧阴囊空虚。四肢肌张力低,双侧巴氏征阳性,膝、跟腱反射可引出。
相关辅助检查:
心脏彩超提示卵圆孔未闭。
头颅、垂体MRI未见明显异常。
阴囊彩超考虑双侧隐睾。
双膝关节侧位片显示髌骨发育不良(E、F)。
甲状腺功能正常。
胰岛素样生长因子<2.5ng/mL(正常参考值:45~305ng/mL)。

全外显子组测序分析结果显示:
患儿的KAT6B第16外显子
c.3367_c.3370delAGAA(p.Arg1123Argfs?6)杂合移码变异,
患儿父母未检测到该变异,该变异为新发变异(de novo)。
临床诊断:
生殖器-髌骨综合征
病例2
患儿,女,19个月,因发育不良被转诊至发育儿科。
父母非血缘关系,无先天异常家族史。产前超声和新生儿筛查均未发现异常,第1胎(G1P0)在胎龄3个月时自然流产。患儿为第2胎第1产,出生时母亲和父亲分别是26岁和27岁。他们的身高分别为150和175cm。
患儿出生时40周,出生体重2850g(z值:−0.7),出生身长48cm(z值:−0.6),头围31.5cm(z值:−2.0)。
出生后,表现出虚弱的哭声和进食困难。母乳喂养,没有呛咳或频繁呕吐。患儿比其他同龄女孩发育慢。人体测量结果如下:根据2006年世卫组织国际生长标准图表进行评估,体重为5.2kg(z值:−2.5),身长为60cm(z值:−3.1),头围为41.2cm(z值:−1.2)。用改良的Gesell发展时间表评估她的发展商(DQ),显示发育迟缓,平均DQ低至52分。
体格检查:
先天性马蹄内翻足。此外,面部畸形表现为眼睑裂隙狭窄、上睑下垂、鼻尖呈球状、宽鼻梁、小口、薄薄的上唇和小而低垂的耳朵(图)。肛门和生殖器没有异常。四肢肌肉张力略高。19个月才能扶站。
心脏超声检查证实房间隔缺损(直径0.36cm)伴有轻度三尖瓣反流。
腹部超声显示左肾4.6cm*2.5cm,右肾5.0cm*2.2cm,都比较小。胰腺饱满(比正常略大)。
骨盆X线示髋臼角大,倾斜,部分髋臼形态浅(左27°,右25°)。
双侧膝关节X线、脑磁共振成像(MRI)和脑电图(EEG)均未见明显异常。
听觉脑干反应(ABR)显示左耳为25dBnHL,右耳为20dBnHL。
X线片中四肢长骨未见明显异常征象。
血液检查包括肾功能、肝功能、电解质和甲状腺功能均未发现异常。
标准核型为46,XX。
考虑生长障碍、全面发育迟缓和临床特征,获得了父母的知情同意进行基因检测。
患儿的KAT6B第16外显子存在变异:

生长发育随访
患儿体格发育和智力发育严重迟缓。
牙齿在9个月大时开始萌出。
11个月时出现翻身。
15个月时出现爬行。
19个月大时,有16颗牙齿。
19个月大时,体重、身高和头围分别为7.8 kg(z值:−1.9)、69cm(z值:−4.2)和44 cm(z值:−1.7)。
19个月大时,仅能扶站,并在支持下迈几步(图D)。
19个月大时,语言仅限于重复使用单元音和辅音音节。
患儿的生长曲线如下图所示:

临床诊断
Say-Barber-Biesecker-Young-Simpson 综合征
病例述评
KAT6B基因位于10q22.2区,该区域包含18个外显子并编码2073个氨基酸,编码的蛋白是一种组蛋白乙酰转移酶和MOZ/MORF蛋白质复合体的组成部分。组蛋白乙酰转移酶可能参与正反转录调控,编码的蛋白质在其N末端的转录激活活性而其C-末端转录抑制活性,这种蛋白质是RUNX2依赖性转录激活的必要条件和可能参与大脑皮层发育是成人神经干细胞中组蛋白H3乙酰转移酶复合物的一部分,对骨骼、神经系统早期发育调节至关重要。
KAT6B基因相关疾病包括
生殖器-髌骨综合征(OMIM606170)和
Say-Barber-Biesecker-Young-Simpson 综合征(SBBYSS)(OMIM603736),均为常染色体显性遗传病。两者共同的表型特征均为显著的全面运动智力发育迟缓、男性生殖器异常(隐睾等)和髌骨发育不全,先天性心脏缺陷、牙齿异常、听力丧失和甲状腺异常也很常见,婴儿期多出现严重的肌张力低下和喂养困难。仅存在于生殖器-髌骨综合征中的特征是髋膝关节的屈曲挛缩、踝关节足部畸形、胼胝体发育不良、肾积水或多发性肾囊肿。仅存在于SBBYSS患者的特征包括长拇指/大拇趾、球状鼻尖、面具脸、眼睑下垂和泪管异常。KAT6B变异的位置与患者的表型相关,这些变异体类型多由插入/缺失和无义变异导致的蛋白质截短,多发生在第16~18外显子。SBBYSS综合征患者变异多发生在第18外显子更远端,缺乏导致功能丧失变异的转录激活结构域,也有研究显示第15、16、17外显子的变异更接近于 SBBYSS的典型特征。SBBYSS性别无特异性,临床表型复杂多样,但KAT6B基因变异所致的生殖器-髌骨综合征和 SBBYSS是临床上类似的疾病,具有一些重叠临床特征,建议将与典型的KAT6B致病变体相关的表型称为“KAT6B谱系障碍”或KAT6B相关疾病”。
根据基因型表型相关性将KAT6B变异分为4组:
第1组变异(密码子1~1205)与轻度 SBBYSS相关;
第2组(密码子121350)与生殖器-髌骨综合征相关;
第3组(密码子13501520)大部分具有生殖器-髌骨/ SBBYSS混合表型;
第4组(密码子1520~2073)几乎完全是 SBBYSS表型。
英文朗读
Say-Barber-Biesecker-Young-Simpson syndrome and genitopatellar syndrome, are characterized by global developmental delay/intellectual disability and special clinical manifestations.
They are two distinct clinically overlapping syndromes caused by truncating sequence variants in the KAT6B (10q22.2) gene.
The patients reported here are mainly characterized by syndromic forms of short stature and developmental delay, which may
contribute to the understanding of clinical genetics for KAT6B-associated disorders.
上面朗读见于配套的视频号:






