singleseqgset | 单细胞RNA-Seq基因集富集分析
singleseqgset是用于单细胞RNA-seq数据的基因集富集分析的软件包。它使用简单的基础统计量(variance inflated Wilcoxon秩和检验)来确定不同cluster中感兴趣的基因集的富集。
Installation
library(devtools)
install_github("arc85/singleseqgset")
library(singleseqgset)
基因集富集分析是一种无处不在的工具,用于从基因组数据集中提取具有生物学意义的信息。有许多不同类型的工具可用于基因集富集分析,以前的基因组富集分析工具的关键概念是比较组内基因与组外基因的差异(例如,各组之间基因表达的log fold change变化)。但最值得肯定的是Subramanian等人(PNAS 2005)(https://www.ncbi.nlm.nih.gov/pubmed/16199517)的开创性工作-Gene Set Enrichment Analysis(GSEA富集分析 - 界面操作)。
GSEA分析中已知功能的基因集可以从许多地方进行获取,包括Broad的Molecular Signatures Database(MSigDB)和The Gene Ontology Resource,关键是选择最能回答当前生物学问题的基因集。举个栗子,通常您不会使用Broad的C7(免疫学基因集)中的基因集来回答有关神经元发育的问题(除非有充分的理由!)。
singleseqgset研究者在这里开发的基因集富集方法灵感来自Di Wu和Gordon K Smyth在Nucleic Acids Research 2012(https://www.ncbi.nlm.nih.gov/pubmed/16199517)上的工作,而Wu和Smyth在这个工作的基础上又开发了相关调整的MEan RAnk基因集测试(CAMERA)。
CAMERA是一项竞争性基因集富集测试,可控制基因集内的基因间相关性。研究团队之所以选择使用CAMERA,是因为它不依赖于基因独立性的假设(因为我们知道基因通常具有结构化的共表达模式),也不依赖于基因标记的排列或测试样品的重采样。CAMERA通过基于基因间相关程度生成方差膨胀因子来控制基因间相关性,并将这种方差膨胀纳入Wilcoxon秩和检验中。
此workflow演示了对模拟数据使用singleseqgset的标准用法。我们将执行以下步骤:
- 使用splatter R包模拟表达式数据;
- 使用msigdbr下载感兴趣的基因集;
- 将特定基因集添加到我们的模拟数据中(就是让我们的模拟数据可以富含某基因集);
- 使用标准的Seurat工作流程(v.2.3.4)处理我们的数据;
- 使用singleseqgset进行基因集富集分析;
- 将结果绘制在热图中;
首先,加载所有必要的程序包,并使用splatter模拟数据。
suppressMessages({
library(splatter)
library(Seurat)
library(msigdbr)
library(singleseqgset)
library(heatmap3)
})
#Splatter是用于模拟单细胞RNA测序count数据的软件包。
#创建参数并模拟数据
sc.params <- newSplatParams(nGenes=1000,batchCells=5000)
sim.groups <- splatSimulate(params=sc.params,method="groups",group.prob=c(0.4,0.2,0.3,0.1),de.prob=c(0.20,0.20,0.1,0.3),verbose=F)
sim.groups #Check out the SingleCellExperiment object with simulated dataset
## class: SingleCellExperiment
## dim: 1000 5000
## metadata(1): Params
## assays(6): BatchCellMeans BaseCellMeans ... TrueCounts counts
## rownames(1000): Gene1 Gene2 ... Gene999 Gene1000
## rowData names(8): Gene BaseGeneMean ... DEFacGroup3 DEFacGroup4
## colnames(5000): Cell1 Cell2 ... Cell4999 Cell5000
## colData names(4): Cell Batch Group ExpLibSize
## reducedDimNames(0):
## spikeNames(0):
colData(sim.groups) #The colData column "Group" contains the groups of simulated cells
## DataFrame with 5000 rows and 4 columns
## Cell Batch Group ExpLibSize
## <factor> <character> <factor> <numeric>
## Cell1 Cell1 Batch1 Group1 73324.2901799195
## Cell2 Cell2 Batch1 Group1 71115.1438815874
## Cell3 Cell3 Batch1 Group3 89689.371004738
## Cell4 Cell4 Batch1 Group3 44896.460028516
## Cell5 Cell5 Batch1 Group3 53956.0668720417
## ... ... ... ... ...
## Cell4996 Cell4996 Batch1 Group2 79041.7558615305
## Cell4997 Cell4997 Batch1 Group4 66684.3797711752
## Cell4998 Cell4998 Batch1 Group3 70407.9613848431
## Cell4999 Cell4999 Batch1 Group3 64645.3257427214
## Cell5000 Cell5000 Batch1 Group3 62266.9119469426
#我们将从sim.groups对象中提取模拟的计数和组别
sim.counts <- assays(sim.groups)$counts
groups <- colData(sim.groups)$Group
names(groups) <- rownames(colData(sim.groups))
table(groups)
## groups
## Group1 Group2 Group3 Group4
## 1977 992 1536 495
有次我们创建了一些模拟数据,这些数据由4个cluster组成,cluster中不同基因的表达比例不同。
Download gene sets of interest using msigdbr在这里,我们将使用misigdbr软件包下载Hallmark Gene Sets。另外,我们可以直接从Broad网站(http://software.broadinstitute.org/gsea/msigdb/index.jsp)下载基因集,然后使用`GSEABase`软件包将其读入R。
注意:必须为您的项目选择适当的基因注释样式(gene symbols或entrez gene ID;在这里我们只是gene symbols)和物种(在这里我们使用human)。否则,您的基因组中的基因名称将与您的数据中的基因名称不匹配,并且所有基因组都将获得“0”富集得分。
h.human <- msigdbr(species="Homo sapiens",category="H")
h.names <- unique(h.human$gs_name)
h.sets <- vector("list",length=length(h.names))
names(h.sets) <- h.names
for (i in names(h.sets)) {
h.sets[[i]] <- pull(h.human[h.human$gs_name==i,"gene_symbol"])
}
我们将分配5个基因集在每个cluster中专门表达(如果不进行分配,则可能出现无功能集在cluster中富集的情况。)。随机选择这20个集合,并从zero-inflated的Poisson分布中抽取每个基因,成功概率等于50%,λ值为10,这将模拟中等dropout和相对较低的基因表达量。
sets.to.use <- sample(seq(1,50,1),20,replace=F)
sets.and.groups <- data.frame(set=sets.to.use,group=paste("Group",rep(1:4,each=5),sep=""))
for (i in 1:nrow(sets.and.groups)) {
set.to.use <- sets.and.groups[i,"set"]
group.to.use <- sets.and.groups[i,"group"]
gene.set.length <- length(h.sets[[set.to.use]])
blank.matrix <- matrix(0,ncol=ncol(sim.counts),nrow=gene.set.length)
rownames(blank.matrix) <- h.sets[[sets.to.use[i]]]
matching <- rownames(blank.matrix) %in% rownames(sim.counts)
if (any(matching==TRUE)) {
matching.genes <- rownames(blank.matrix)[matching]
nonmatching.genes <- setdiff(rownames(blank.matrix),matching.genes)
blank.matrix <- blank.matrix[!matching,]
sim.counts <- rbind(sim.counts,blank.matrix)
} else {
sim.counts <- rbind(sim.counts,blank.matrix)
matching.genes <- rownames(blank.matrix)
}
group.cells <- colnames(sim.counts)[groups==group.to.use]
for (z in group.cells) {
if (any(matching==TRUE)) {
sim.counts[matching.genes,z] <- ifelse(rbinom(length(matching.genes),size=1,prob=0.5)>0,0,rpois(length(matching.genes),lambda=10))
sim.counts[nonmatching.genes,z] <- ifelse(rbinom(length(nonmatching.genes),size=1,prob=0.5)>0,0,rpois(length(nonmatching.genes),lambda=10))
} else {
sim.counts[matching.genes,z] <- ifelse(rbinom(length(matching.genes),size=1,prob=0.5)>0,0,rpois(length(matching.genes),lambda=10))
}
}
}
#Here, we will check out the sum of expression for the first gene set
len.set1 <- length(h.sets[[sets.to.use[[1]]]])
plot(apply(sim.counts[1001:(1000 len.set1),],2,sum),xlim=c(0,5000),xlab="Cells",ylab="Sum of Gene Set 1 Expression")

我们已经成功地修改了模拟计数矩阵,使其包含特定cluster中感兴趣的基因集。例如,我们知道Group1细胞应显示出以下基因的富集:
HALLMARK_APICAL_SURFACE, HALLMARK_COMPLEMENT, HALLMARK_DNA_REPAIR, HALLMARK_IL2_STAT5_SIGNALING, HALLMARK_UNFOLDED_PROTEIN_RESPONSE。
Seurat workflow on simulated data
#构建seurat object
ser <- CreateSeuratObject(raw.data=sim.counts)
#归一化
ser <- NormalizeData(ser)
ser@var.genes <- rownames(ser@raw.data)[1:1000]
ser <- ScaleData(ser,genes.use=ser@var.genes)
## Scaling data matrix
#我们会假设1000个模拟基因是“可变基因”,
#我们将跳过Seurat的FindVariableGenes
ser <- RunPCA(ser,pc.genes=ser@var.genes,do.print=FALSE)
PCElbowPlot(ser)

#其中top4PCs代表了多数的差异
#我们将会选择top5 PCs;
ser <- RunTSNE(ser,dims.use=1:5)
#将模拟的dataID号加入Seurat object
data.to.add <- colData(sim.groups)$Group
names(data.to.add) <- ser@cell.names
ser <- AddMetaData(ser,metadata=data.to.add,col.name="real_id")
ser <- SetAllIdent(ser,id="real_id")
#我们将会跳过使用Seurat聚类的步骤,因为我们已知道真正的聚类IDs
TSNEPlot(ser,group.by="real_id")

首先,我们将基于logFC计算基因集富集测试的矩阵。我们选择使用已针对library大小进行校正的log-normalized数据,并存储在Seurat的“@data”slot中。
logfc.data <- logFC(cluster.ids=ser@meta.data$real_id,expr.mat=ser@data)
names(logfc.data)
## [1] "cluster.cells" "log.fc.cluster"
logFC函数返回一个长度为2的列表,其中包含每个cluster中的细胞以及cluster之间基因的对数倍变化。下一步需要此数据计算enrichment scores和p值。
gse.res <- wmw_gsea(expr.mat=ser@data,cluster.cells=logfc.data[[1]],log.fc.cluster=logfc.data[[2]],gene.sets=h.sets)
## [1] "Removed 1 rows with all z-scores equal to zero."
names(gse.res)
## [1] "GSEA_statistics" "GSEA_p_values"
此函数返回两个列表,第一个包含测试统计信息“GSEA_statistics”,第二个包含p值“GSEA_p_values”。我们可以将这些结果绘制为热图,以可视化差异调节基因集。
res.stats <- gse.res[["GSEA_statistics"]]
res.pvals <- gse.res[["GSEA_p_values"]]
res.pvals <- apply(res.pvals,2,p.adjust,method="fdr") #Correct for multiple comparisons
res.stats[order(res.stats[,1],decreasing=TRUE)[1:10],] #Top gene sets enriched by z scores
## Group1 Group2 Group3
## HALLMARK_IL2_STAT5_SIGNALING 10.51698615 -7.55892580 -8.113191
## HALLMARK_DNA_REPAIR 10.47159479 -7.62264059 -7.326454
## HALLMARK_COMPLEMENT 10.17982979 -3.68698969 -8.347697
## HALLMARK_UNFOLDED_PROTEIN_RESPONSE 9.54278550 -8.98321228 -7.275756
## HALLMARK_APICAL_SURFACE 7.63384635 -5.70116613 0.000000
## HALLMARK_ANGIOGENESIS 1.32886963 -0.91447563 0.000000
## HALLMARK_HEDGEHOG_SIGNALING 0.77628519 0.66226233 0.000000
## HALLMARK_KRAS_SIGNALING_UP 0.59844933 -0.20261236 -2.365948
## HALLMARK_COAGULATION 0.57787404 9.03118414 -6.988972
## HALLMARK_INFLAMMATORY_RESPONSE 0.05206778 -0.08661822 -1.260939
## Group4
## HALLMARK_IL2_STAT5_SIGNALING -8.4400626
## HALLMARK_DNA_REPAIR -10.3905166
## HALLMARK_COMPLEMENT -12.5780500
## HALLMARK_UNFOLDED_PROTEIN_RESPONSE -5.8693210
## HALLMARK_APICAL_SURFACE 0.0000000
## HALLMARK_ANGIOGENESIS -0.6935659
## HALLMARK_HEDGEHOG_SIGNALING 0.3350711
## HALLMARK_KRAS_SIGNALING_UP -1.1822606
## HALLMARK_COAGULATION -4.3269207
## HALLMARK_INFLAMMATORY_RESPONSE -3.3347607
res.pvals[order(res.stats[,1],decreasing=TRUE)[1:10],] #Top gene sets by p values
## Group1 Group2 Group3
## HALLMARK_IL2_STAT5_SIGNALING 2.858190e-24 2.489271e-13 3.451510e-15
## HALLMARK_DNA_REPAIR 2.858190e-24 1.739767e-13 1.286641e-12
## HALLMARK_COMPLEMENT 3.985134e-23 6.949503e-04 6.821492e-16
## HALLMARK_UNFOLDED_PROTEIN_RESPONSE 1.362655e-20 2.577150e-18 1.687982e-12
## HALLMARK_APICAL_SURFACE 1.594960e-13 5.830539e-08 1.000000e 00
## HALLMARK_ANGIOGENESIS 3.003553e-01 5.697704e-01 1.000000e 00
## HALLMARK_HEDGEHOG_SIGNALING 5.642487e-01 7.318339e-01 1.000000e 00
## HALLMARK_KRAS_SIGNALING_UP 6.589077e-01 1.000000e 00 4.406075e-02
## HALLMARK_COAGULATION 6.589077e-01 2.080345e-18 1.130707e-11
## HALLMARK_INFLAMMATORY_RESPONSE 1.000000e 00 1.000000e 00 3.631134e-01
## Group4
## HALLMARK_IL2_STAT5_SIGNALING 1.942653e-16
## HALLMARK_DNA_REPAIR 6.709712e-24
## HALLMARK_COMPLEMENT 1.366256e-34
## HALLMARK_UNFOLDED_PROTEIN_RESPONSE 2.144160e-08
## HALLMARK_APICAL_SURFACE 1.000000e 00
## HALLMARK_ANGIOGENESIS 6.462100e-01
## HALLMARK_HEDGEHOG_SIGNALING 7.856739e-01
## HALLMARK_KRAS_SIGNALING_UP 3.417063e-01
## HALLMARK_COAGULATION 4.939474e-05
## HALLMARK_INFLAMMATORY_RESPONSE 2.460747e-03
names(h.sets)[sets.to.use[1:5]] #Compare to the simulate sets we created
## [1] "HALLMARK_APICAL_SURFACE"
## [2] "HALLMARK_COMPLEMENT"
## [3] "HALLMARK_DNA_REPAIR"
## [4] "HALLMARK_IL2_STAT5_SIGNALING"
## [5] "HALLMARK_UNFOLDED_PROTEIN_RESPONSE"
#Plot the z scores with heatmap3
heatmap3(res.stats,Colv=NA,cexRow=0.5,cexCol=1,scale="row")
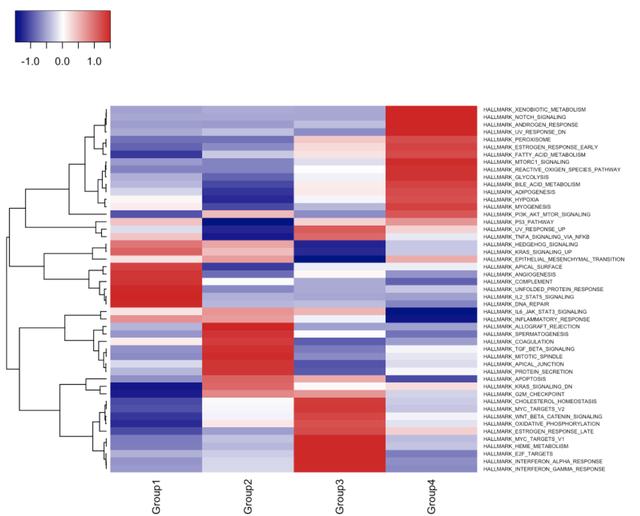
恭喜,您已经使用singleseqgset执行了第一个基因集富集分析!
,




