*今日头条上无法显示上标、下标,欲获得更好的阅读体验请前往微信公众号。
*前往“纳米酶Nanozymes”公众号,了解有关纳米酶的最新消息!
*本文首发于“纳米酶Nanozymes”公众号,2021年7月15日
*编辑:俞纪元
引言
大家好,今天分享一篇发表在Chem. Sci.上的文章<Bound oxygen-atom transfer endows peroxidase-mimic M-N-C with high substrate selectivity>。本文通讯作者是东南大学化学化工学院张袁健教授,他们组(https://Zhang.team)的主要研究方向是Carbosensing-基于富碳材料的分子传感。

结合态氧原子转移机制:
M-N-C选择性催化氧化类过氧化物酶底物
文章简介
数万年的自然演化赋予了天然酶高活性、高底物选择性的精巧结构,然而其分离/纯化成本高昂,极端条件下易失活。为此,纳米酶作为一类既有纳米材料理化特性,又蕴含酶学催化功能的新一代人工模拟酶应运而生,其中,如何提高纳米酶的底物选择性是该领域面临的巨大挑战和重要科学问题。本文以生命分析化学中的两个重要反应3,3',5,5'-四甲基联苯胺(TMB)和3-氨基邻苯二甲酰肼(Luminol)的催化氧化为研究模型,发现通过调节氮配位过渡金属掺杂碳(M-N-C)的金属配位中心可将其类过氧化物酶底物选择性显著提高至200倍。机理研究表明,在N配位金属位点形成结合态氧中间体(M=O)是H2O2连续活化及随后底物以高选择性方式氧化的关键,并且嵌于刚性石墨结构的N配位金属位点较天然辣根过氧化物酶(HRP)具有更高的底物选择性。这种结合态中间体机制与常见的自由态活性氧中间体(ROS)机制截然不同,对于开发高底物选择性的纳米酶具有重要的启示作用。
图文解析
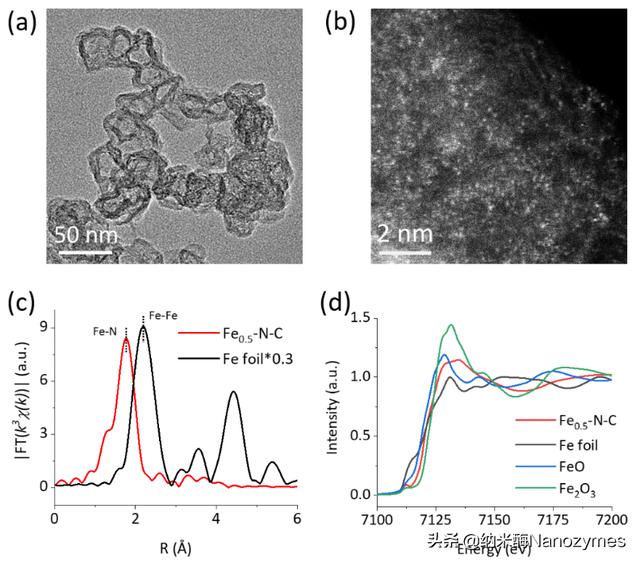
图1 M-N-C纳米结构及分子结构表征:Fe-N-C纳米酶的(a)TEM及(b)HAADF-STEM图像;Fe-N-C及参考样品在Fe K-edge的(c)FT-EXAFS图谱及(d)XANES图谱
M-N-C纳米酶通过经典的高温热解结合酸蚀刻的方法制得,TEM结果(图1a)表明,尺寸约50 nm的无规则纳米颗粒状Fe-N-C主要由两部分组成,一是载体炭黑(CB)呈涡轮状的多层石墨结构;二是邻苯二胺聚合物热解生成的包覆于CB的片层碳,并且未从TEM图像中观察到任何金属晶相。分辨率可达亚埃级的HAADF-STEM研究证明了Fe-N-C纳米酶中的金属以单原子形式存在且均匀分布于碳基底(图1b)。Fe K-edge的XAFS谱进一步探究了Fe-N-C纳米酶中Fe的配位结构和价态,Fe-N-C和Fe箔的FT-EXAFS图谱如图1c所示,Fe箔在2.19Å处呈现出明显Fe-Fe峰,而Fe-N-C纳米酶不存在该峰,进一步证明了Fe-N-C纳米酶中的Fe以原子级形式存在;另外Fe-N-C纳米酶位于1.76 Å处的主峰证明了存在Fe-N配位。从XANES图谱(图1d)可见,Fe-N-C的吸收边位于FeO和Fe2O3的吸收边之间,表明Fe-N-C中Fe的价态介于 2与 3之间。Fe-N-C的其他结构信息及其他M-N-C(M = Mn、Co、Ni、Cu)的结构特征见原文。

图2 M-N-C、N-C、CB及HRP催化类过氧化物酶反应的底物选择性研究:(a)催化TMB氧化的反应方程式及M-N-C、N-C、CB、HRP催化TMB氧化显色的照片;(c)20 mg mL-1 M-N-C、N-C及CB在0.1 M HAc-NaAc水溶液(pH 3.6)中催化100 mM H2O2氧化1 mM TMB的反应初速率;(b)Luminol催化氧化的反应方程式及M-N-C、N-C、CB、HRP催化luminol氧化发光的照片;(d)50 μg mL-1 M-N-C、N-C及CB在0.01 M NaOH水溶液中催化250 mM H2O2氧化2.5 mM luminol的最强发光强度(425 nm)
H2O2氧化TMB与luminol是类过氧化物酶性质研究的两个经典反应模型,本文以这两个模型反应为案例,探究了M-N-C催化类过氧化物酶反应的底物选择性。首先,本文研究了M-N-C、N-C及CB在HAc-NaAc水溶液(pH 3.6)中催化H2O2氧化TMB的性能,如图2a所示,除CB外,M-N-C及N-C均可催化TMB氧化产生蓝色的氧化产物,且Fe-N-C催化反应所得颜色较其他更深,因此Fe-N-C活性最为突出。进一步通过紫外可见吸光度法测试了M-N-C、N-C及CB催化TMB氧化产物在652 nm处的动力学曲线,计算得到了该反应的初速率(图2c),可见Fe-N-C纳米酶的催化速率可达6.08 μM s-1,远高于其他纳米酶,尤其是Co-N-C。
进一步通过检测luminol化学发光(CL)强度研究了M-N-C、N-C、CB及HRP在NaOH溶液中催化H2O2氧化luminol的活性。由图2b中照片可见,仅在Co-N-C和HRP催化反应中出现了明显的肉眼可见的CL信号。通过光纤光谱仪的时序图程序测定催化过程中luminol在425 nm处的特征发射峰的动力学曲线可量化CL强度(图2d),同样,区别于Co-N-C,其他M-N-C、N-C及CB未检测到明显的光信号。由于M-N-C具有类似的粒子尺寸、形貌及碳结晶度,因此作者推测M-N配位中心对于类过氧化物酶底物选择性催化起着至关重要的作用。

图3 (a)ROS清除剂对Fe-N-C催化H2O2氧化TMB的影响;(b)Fe-N-C H2O2 ROS自旋捕获剂的ESR图谱;(c)HRP H2O2 ROS自旋捕获剂的ESR图谱;(d)110 K测得的Fe-N-C及120 ℃真空脱气处理36 h所得Fe-N-C的ESR图谱
鉴于TMB及luminol均是可被H2O2活化生成的活性氧自由基(ROS)氧化的底物,为了探究Fe-N-C及Co-N-C催化H2O2氧化这两个底物的内在机制,本文首先通过自由基清除实验研究了H2O2活化过程产生的自由基类型。以Fe-N-C催化TMB氧化反应为例,如图3a所示,当催化体系中分别加入超氧化物歧化酶(SOD)及甘露醇(Mannitol)以清除反应中可能产生的超氧及羟基自由基时,未观察到该催化反应活性受影响,因此推测该反应不涉及这两种自由基。随后本文进一步通过电子自旋共振(ESR)图谱研究了反应涉及的ROS(图3b),同样证明Fe-N-C催化活化H2O2过程中没有产生单线态氧、超氧及羟基自由基,该过程与HRP催化机制类似(图3c)。原位监测反应过程中涉及的Fe=O的生成与消耗对于研究Fe-N-C催化H2O2氧化TMB的机制至关重要但也存在巨大挑战,本文通过非原位的方法研究了Fe-N-C与含氧基团之间存在相互作用,如图3d所示,Fe-N-C经真空加热处理后,Fe的特征峰明显减弱,侧面反应了Fe与O之间存在相互作用。

图4 (a)TMB氧化体系中Fe-N-C及Co-N-C催化第一分子H2O2的能量图及相应的反应构型;(b)luminol氧化体系中Fe-N-C及Co-N-C催化第一分子HO2-的能量图及相应的反应构型;其中IS、MS及TS分别指代初始态、中间态及过渡态
TMB氧化体系中,Fe-N-C及Co-N-C催化第一分子H2O2的能量图及相应的反应构型如图4a所示,H2O2吸附于Fe-N-C、Co-N-C的Eads分别为-0.16 eV和-0.13 eV,说明H2O2吸附于Fe-N-C较Co-N-C略稳定;从过渡态构型可见,随后在Fe-N-C及Co-N-C作用下H2O2均发生了O-O断键,并且Fe-N-C体系的活化能(Ea)很低(0.08 eV),而Co-N-C体系的Ea相对较高(0.94 eV),因此Fe-N-C相较于Co-N-C更易催化O-O断键,这可能是导致二者活性巨大差异的核心因素。
Luminol氧化体系中,Fe-N-C及Co-N-C催化第一分子HO2-的能量图及相应的反应构型如图4d所示,HO2-吸附于Fe-N-C、Co-N-C的Eads分别为-1.38 eV和-1.18 eV,说明HO2-吸附于Fe-N-C较Co-N-C略稳定;从过渡态构型可见,随后HO2-在Fe-N-C作用下发生了O-O断键,而在Co-N-C作用下O-O键明显变长,并且Fe-N-C催化HO2-的Ea(1.64 eV)明显高于Co-N-C体系的Ea(0.81 eV),因此Co-N-C相较于Fe-N-C更易活化HO2-产生M=O。

图5 (a)Fe-N-C催化H2O2氧化TMB的反应机理;(b)Co-N-C催化HO2-氧化TMB的反应机理
因此,论文作者提出了M-N-C类过氧化物酶底物选择性的机理。简而言之,在TMB氧化体系中,H2O2首先吸附于Fe-N-C中N配位的FeIII;然后,被吸附H2O2发生O-O断键,FeIII氧化至FeV=O并释放出H2O分子;后续是否发生底物氧化抑或H2O2歧化取决于二者的供电子能力大小。同样,在luminol氧化体系中,HO2-首先吸附于Co-N-C中N配位CoII;然后,被吸附HO2-发生O-O断键,CoII氧化至CoIV=O并释放出OH-;后续是否发生底物氧化抑或HO2-歧化取决于二者的供电子能力大小。更多信息参见原文。
结论与展望
综上所述,本文提出通过改变M-N-C活性中心的金属类型可以实现催化底物选择性氧化,该选择性源于反应过程中在氮配位过渡金属上形成了结合态氧中间体(M=O)。以Fe-N-C和Co-N-C为例,得益于二者氮配位金属中心的d轨道差异,其催化TMB与luminol氧化的选择性高达200倍;并且,M-N-C在催化类过氧化物酶反应中表现出的高底物选择性甚至优于天然酶HRP,这可能由于M-N-C的金属配体嵌于刚性石墨骨架所制。实验和基于简化模型的DFT计算均证明,M-N配位中心是M-N-C催化类过氧化物酶底物选择性氧化的来源,决定活化H2O2的能力强弱及生成后续氧转移反应的M=O活性中间体;另外,底物与M=O活性中间体作用的结合能及其供电子能力也决定着反应选择性。值得注意的是,结合态M=O中间体催化机制与已报道的大多数类过氧化物酶的游离ROS中间体机制完全不同,这对开发下一代具有高底物选择性纳米酶具有重要的指导意义。此外,这项工作还强调了借鉴天然酶催化机制并结合纳米材料结构特征可模拟并在某些方面超越天然酶的功能,进而实现生物学所无法完成的更高级应用。
原文:Xinghua Chen, Lufang Zhao, Kaiqing Wu, Hong Yang, Qing Zhou, Yuan Xu, Yongjun Zheng, Yanfei Shen, Songqin Liu, Yuanjian Zhang*, Bound Oxygen-Atom Transfer Endowing Peroxidase-Mimic M-N-C with High Substrate Selectivity. Chem. Sci. 2021, 12, 8865-8871 (Chem Sci Pick of the Week).
撰稿:陈兴华 审阅:王权
编辑:周逸夫
,




