文章背景
含锰材料可以提高电池可持续性,并降低大规模生产电池的成本。然而,由于活性Mn中的Jahn–Teller(J–T)畸变和电池运行过程中不可逆的结构变化之间的相互作用,这些材料中的大多数都会发生降解。许多降解过程起源于电化学界面,并在纳米尺度上相互连接。探究不同界面过程之间的关系(即颗粒与颗粒和颗粒与电解质的关系)一直以来都是电池研究的挑战。
在商用非水锂离子电池中,非水溶剂和空气敏感盐的高蒸气压妨碍了对于界面的观测。因此,大多数先前的研究都使用非原位表征来探测循环后的电极表面,缺乏时间分辨率。此外,原位技术,如透射电子显微镜(TEM)显示出优异的时间和空间分辨率,但通常缺乏对复杂和异质电极表面的统计特异性。因此,很少考虑颗粒和电极的不均匀性,尽管它们在降解机制和电池性能中发挥着重要作用。因此,迫切需要对锰基正极进行多维研究,以从时间和空间分辨率上统计阐明正极表面的动力学和降解机制。
内容简介
在这项研究中,本文可视化并量化了工作状态下含锰正极的时间和空间分辨的锰溶解/再沉积(D/R)动力学。粒子级和电极级分析表明,在不同的电荷状态下,D/R动力学与不同的界面降解机制有关。作者在统计上区分了表面重建和Jahn–Teller畸变对不同工作电压下Mn溶解的贡献。将磺化聚合物(Nafion)引入复合电极中可以通过捕获溶解的Mn物种和快速建立局部Mn D/R平衡来调节D/R动力学。首次通过操作同步加速器研究来确定导致Mn溶解的化学和结构转变的,并开发了一种调节Mn界面动力学以提高电池性能的有效方法。相关成果以“Operando characterization and regulation of metal dissolution and redeposition dynamics near battery electrode surface ”为题发表在国际期刊nature nanotechnology 上。文章通讯作者是林峰,一作是Yuxin Zhang。
文章亮点
1、本工作揭示了D/R动态的实时可视化和量化,并进行了单粒子分辨率和电尺度的统计分析。
2、存在于活性粒子表面的磺化四氟乙烯(即Nafion)与溶解的Mn阳离子强烈配位,降低质子导电性,并改善电池动力学和循环寿命。
3、本研究为调控可充电电池电化学界面上的Mn D/R动态提供了新见解。
主要内容
本文选择2M双(三氟甲磺酰基)酰亚胺锂(LiTFSI)水性电解质来研究LMO电极中的Mn D/R动力学。这种稀释的电解质可以在短时间内将Mn的溶解加速到可量化的浓度。图1a显示了LMO的循环伏安法(CV)数据,具有对应于Mn3 /Mn4 氧化还原对的峰。上截止电压附近的高电流密度是由Mn氧化反应和析氧反应(OER)共同贡献的。材料退化可以通过在15个循环内逐渐衰减的峰值电流来观察。然后,应用原位X射线荧光显微镜(XFM)来跟踪整个电化学循环过程中电极水平上的Mn分布。原始的液滴制备LMO电极显示出不均匀的Mn分布(图1b),具有一些聚集区域。
由于Mn的溶解,连续的CV循环降低了高浓度区域的强度,并降低了Mn分布的总体不均匀性。然后,对每个XFM图的数据采集时间进行编程,使其与每个CV循环所需的时间相同,这能够跟踪每个循环后的净Mn浓度变化。观察到,Mn浓度在前十个循环内表现出单调下降,然后在接下来的循环中变得稳定(图1c)。这里给出的净浓度变化反映了Mn D/R过程有利于溶解,直到在十个CV循环后达到平衡。随后,对每个XFM图的数据采集时间进行编程,使其比每个CV周期所需的时间更长,这使得每个XFM图可覆盖一个以上的完整CV周期。Mn浓度表现出非单调波动(图1d),这表明了依赖于电压的D/R行为。
事实上,当我们在不同电位下进行计时电流法(CA)测量时,Mn浓度在电位(0.9 V)时降低,在电位(-0.1 V)时增加。作者进一步在相邻图之间进行逐像素的数学减法,以获得相邻CV循环之间的空间分辨Mn浓度变化。负和正浓度变化分别对应于相邻图谱之间的Mn净浓度损失(溶解>再沉积)和净浓度增益(再沉积>溶解)(图1e)。第二次和第三次循环之间的相减产生了更宽、更负移的直方图,表明Mn D/R更不均匀,并且在早期循环期间存在净Mn浓度损失。相比之下,第十四和第十五周期之间的相减产生了以0%相对浓度变化为中心的较窄直方图。两种减法之间的比较进一步证明了早期循环期间明显的溶解行为,与图1c中的数据一致。
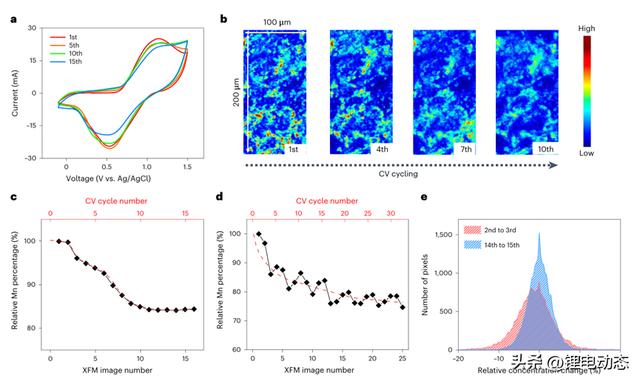

图2. 取决于电压的Mn溶解行为。a)LMO电极在2 M LiTFSI水性电解质中的充放电曲线,其中电压范围为0–1.2 V,相对于Ag/AgCl,电流密度为1 C(100 mA g–1)。b)原位XFM测量的第一个充电曲线(~0.8 C)和不同充电电压下相应的颗粒水平Mn浓度图。像素大小为300 nm×300 nm,图像大小为12μm×15μm。c)根据b.d中的结果计算的颗粒级Mn损失率,用于电极级分析的原位XFM测量的第一个充电曲线(~0.8 c),其中电压保持恒定在1.20和1.55V,用于额外的XFM测量。蓝色、黄色和粉色背景色分别代表LV、MV和HV范围。e)充电过程中电极水平Mn损失率。电压保持过程从标记为1.20和1.55V的数据点开始。f)在MV和HV范围内对Mn D/R动力学进行逐像素分析。
为了揭示不同电压范围下锰的溶解机理,应用原位硬X射线吸收光谱(XAS)研究了LMO在循环过程中的性质。选择了五种SOC,即原始状态、1.00 V、1.20 V、1.45 V和放电状态(图3a)。基于参考和LMO样品的白线能量,表明Mn从原始状态下的3.5 连续氧化到1.2V充电状态下的3.9 ,而它在HV区域的变化可以忽略不计(图3b–d)。扩展X射线吸收精细结构(EXAFS)(图3e)表明,当LMO进入HV范围时,Mn–O和Mn–Mn原子间距离经历异常伸长(图3f)。EXAFS拟合结果显示,相应的Mn–O配位数从6个减少到5个(图3g)。
这些结果共同揭示了由HV下的OER引起的LMO的结构降解,其源于晶格氧损失。1.55V带电LMO颗粒的Mn L2,3电子能量损失谱(EELS)表现出蓝移(图3h,i)从表面向本体移动,证实Mn在表面上被还原。软XAS结果表明,LMO在充电至1V时经历典型的氧化过程(图3j)。然而,在充电到1.20V(MV)和1.55V(HV)时,表面的Mn变得减少,这与EELS结果一致,并证实了表面重建的出现。

图3.电化学循环过程中LMO的化学和结构转变。a)现场硬XAS测量的充放电曲线,其中电流密度为0.2 C(20 mA g–1)。b、 c)不同SOC处的Mn K边缘硬XAS光谱(b)和白线区域(c)的相应放大视图。d)作为SOC函数的白线能量。e)不同SOC下Mn K边EXAFS的傅立叶变换幅度。f)不同SOC下的原子间距离。g)Mn–O和Mn–Mn的配位数是从Mn K边缘EXAFS模拟中获得的。误差条是在EXAFS模拟误差的基础上生成的。h)扫描路径的HAADF-STEM图像。i)沿着扫描路径的MnL2,3-边缘EELS光谱。j)LMO电极在不同SOC下的Mn L3边缘软XAS光谱。例如,1.55 VC表示充电至1.55 V的电极,0 VDC表示放电至0 V的电极。使用表面敏感TEY模式进行测量。
由于LMO的容量小于理论值(148 mAh g–1),并且硬XAS结果(图3g)表明Mn在1.55 V下未完全氧化,即使在HV范围内,许多粒子仍保持部分带电,这表明电极水平上的电荷分布不均匀(具有不同颜色的粒子;图4),这与颗粒水平上不同的Mn D/R行为有关。
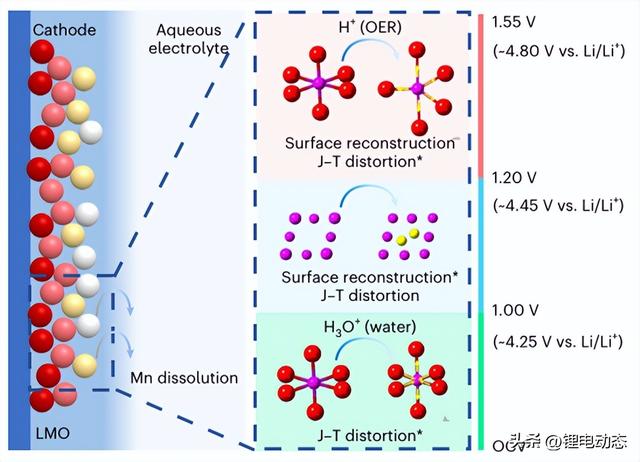
图4.LMO在2M LiTFSI水性电解质中的Mn溶解机制。取决于电压的Mn溶解机制示意图,其中星号表示主要降解驱动力。LMO颗粒的不同颜色表示不同的SOC,其中红色表示最高SOC,而白色表示最低SOC。与远离集电器的颗粒相比,靠近集电器的粒子表现出更高的SOC。黄色箭头表示粘结长度的变化。OCV代表开路电压。
本文发现在电极制备过程中简单地添加Nafion可以有效地减轻复合电极在循环过程中的容量损失。如图5a所示,添加Nafion的LMO的容量保持率得到了提高。与纯LMO相比,添加Nafion的LMO电极在LV范围内表现出较小的反应过电位和较高的输出容量(图5b)。在低电压下,Mn3O4中的Mn2 可以很容易地溶解到电解质中,从而在原位XFM测量过程中,在LV范围内引起更多的Mn损失(图5c,d)。相反,在MV范围(1.0–1.2 V)下,Nafion层可以改变LMO和水性电解质之间的相互作用,并抑制Mn的溶解。重要的是,添加Nafion的LMO电极在第四次充电时表现出可忽略不计的净Mn损失。如图5e,这种现象可能归因于Nafion中的磺酸根可以与带正电的Mn离子缔合,并减少质子跳跃位点,这有效地减少了质子传输,并防止了表面受到质子的攻击。

图5.添加Nafion的LMO材料的电化学性能和潜在的Mn溶解行为。a)添加Nafion的LMO电极在2 M LiTFSI水性电解质中的充放电曲线,其中电压范围为0–1.2 V,相对于Ag/AgCl,电流密度为1 C(100 mA g–1)。b)纯LMO和Nafion添加的LMO的第一充电曲线的比较,其中Nafion加入的LMO在充电时显示出较小的过电势。c)Nafion添加的LMO在不同SOC下的原位XFM映射,其中色标表示Mn浓度。像素大小为2μm×2μm,图像大小为100μm×100μm。d)纯LMO和Nafion添加的LMO电极在充电时的Mn损失。e)第四次充电过程中Nafion添加的LMO颗粒周围的化学环境示意图。
结论
使用原位XFM和一套表面和体相敏感技术,本文对水性电解质中的Mn D/R动力学和相应的LMO降解机制有了全面的了解。连续的Mn溶解和增加的无活性LMO结构域共同导致循环时的性能衰减。J–T畸变和表面重建是电压依赖性Mn溶解行为的原因。Mn的溶解表现出与电压的非线性关系,并且当进行表面重建时可以观察到更高的速率。在电极中引入Nafion可以促进反应动力学并抑制Mn的溶解,特别是由表面重建引起的溶解。同时,Nafion中捕获的Mn2 可以阻碍质子向LMO颗粒表面扩散,并加速Mn D/R平衡的建立,从而提高稀释电解质中的循环寿命。本研究直接可视化和量化了过渡金属的D/R动力学,并揭示了磺化聚合物在控制D/R动力学和提高电池性能方面的作用。本文开发的原位实验方法可以广泛应用于研究其他涉及固液界面的电化学系统。
参考文献
Yuxin Zhang, Anyang Hu, Dawei Xia, Sooyeon Hwang, Sami Sainio, Dennis Nordlund, F. Marc Michel, Robert B. Moore, Luxi Li & Feng Lin. Operando characterization and regulation of metal dissolution and redeposition dynamics near battery electrode surface,nature nanotechnology, 2023.
https://doi.org/ 10.1038/s41565-023-01367-
,




