全基因组关联分析 (Genome-wide association studies, GWAS)现已成功地在复杂的人类特质(traits)和疾病当中应用,例如身高和精神分裂症,辨别出相关的单核苷酸多态性 (SNP)。这种研究方法是在人类基因组范围内找到序列变异,从而筛选出与特质或疾病相关的序列。
GWAS推动了我们对复杂疾病的认知,但由于1)GWAS需要大量的数据和被试来提高它的统计效能,2)难以从生物学的角度解释GWAS结果,这给疾病基因的功能验证和治疗方案的提出带来挑战(1)。
近几年来,大规模的项目例如Genotype-Tissue Expression(GTEx)的开展,给我们带来大量的人类组织基因型和基因表达相匹配的数据(2,3)。这些项目给我们提供了途径,从而探索跨组织转录调节机制和辨别更多的表达数量性状基因座(expression quantitative trait loci, eQTL,3,4) 。同时,这也为我们提供了平台,从而能够解释SNPs,基因和复杂特质之间的联系(5,6,7)。
例如,通过eQTL和GWAS结果的匹配,能够辨别复杂特质相关的风险基因,而其中一种方法,研究者把它称为全转录组关联分析 (transcriptome-wide association analysis, TWAS)。
这种方法首先是在训练后的插补模型 (imputation model)基础上,使用GWAS的基因型数据,对基因表达的数据进行插补(8,9)。其次,在插补后的基因表达和复杂特质之间对基因层面的关系进行评估。这种研究方法让我们增强了对许多疾病和复杂特质遗传基础的认知(10,11)。
图一:2014年发表在Nature上关于精神分裂症相关位点的曼哈顿图 (13)
哈佛的团队利用TWAS整合了精神分裂症患者的GWAS数据和来自大脑,血液和脂肪组织的基因表达数据。在鉴定出的157个精神分裂症相关基因中,35个基因是之前并未发现的,且42个基因和染色质特质(chromatin features)相关,为接下来的研究指明了方向。
而需要指出的是,此项研究运用的方法(见图二))也有一些弊端。例如,由于转录调解取决于选择的人类组织,现在的插补模型均基于不同的人类组织。同时, 对于一些复杂特质,我们并不清楚与其生物相关联的人类组织。最后,挑战还在于难以在有限的数据中,针对任何单一的人类组织做出准确的插补模型。

图二:2018年哈佛团队对精神分裂症的TWAS研究方法
针对这些问题,最近,耶鲁Hongyu zhao的团队提出了一个新的框架去分析TWAS:UTMOST。相比较传统的TWAS的插补模型建立在分别的人类组织上,这个方法整合了44种人类组织,从而允许跨组织(cross-tissue)基因表达的插补和基因层面的相关分析(见图三的UTMOST的分析步骤)。
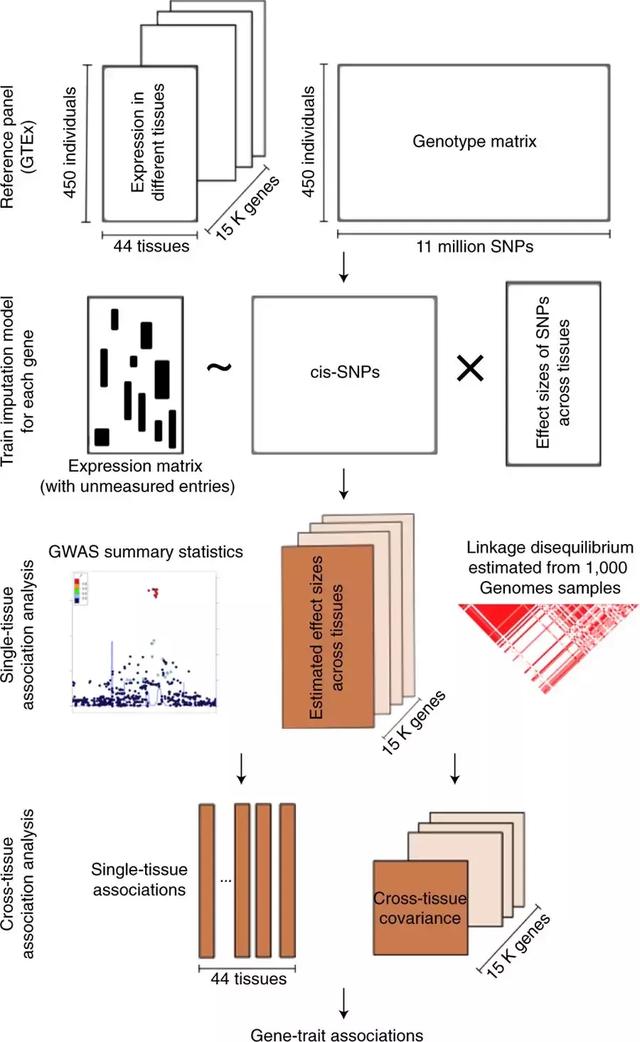
图三:UTMOST的分析步骤
他们证实了这种方法能够提高在人类组织中基因表达数据插补的准确性,并且UTMOST能够在之后的下游关联分析(downstream association analysis)中提供一个强有力的标准,从而可以总结跨组织的基因层面相关分析,并可以整合不同的分子表型(molecular phenotypes)。例如,他们利用UTMOST在50种复杂人类特质中,辨别出更多和人类组织有关的基因(图四)。

图四:UTMOST在50中复杂特质中辨别出更多和人类组织有关的基因
将其和传统的TWAS研究方法比较(图二:传统的TWAS;图三:UTMOST),UTMOST为我们提供了一个更为灵活的基因层面的相关分析,这有利于研究者研究复杂特质和疾病,例如精神分裂症和癌症。
UTMOST software, https://github.com/Joker-Jerome/UTMOST/
主要参考文献:2019年2月25日发表在nature genetics杂志:A statistical framework for cross-tissue transcriptome-wide association analysis其他参考文献:
1.Boyle, E. A., Li, Y. I. & Pritchard, J. K. An Expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177–1186 (2017).
2. Ardlie, K. G. et al. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
3. Aguet, F. et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
4. Yang, F. et al. Identifying cis-mediators for trans-eQTLs across many human tissues using genomic mediation analysis. Genome Res. 27,1859–1871 (2017).
5. Mohammadi, P., Castel, S. E., Brown, A. A. & Lappalainen, T. Quantifying the regulatory effect size of cis-acting genetic variation using allelic fold change. Genome Res. 27, 1872–1884 (2017).
6. Nicolae, D. L. et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS. Genet. 6, e1000888 (2010).
7. Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
8. Gamazon, E. R. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 47, 1091–1098 (2015).
9. Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016).
10. Mancuso, N. et al. Integrating gene expression with summary associationstatistics to identify genes associated with 30 complex traits. Am. J. Hum. Genet. 100, 473–487 (2017).
11. Hoffman, J. D. et al. Cis-eQTL-based trans-ethnic meta-analysis reveals novel genes associated with breast cancer risk. PLoS Genet. 13, e1006690 (2017).
12. Gusev, A., Mancuso, N., Won, H., Kousi, M., Finucane, H. K., Reshef, Y., ... & Ophoff, R. A. (2018). Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nature genetics, 50(4), 538.
13. Ripke, S., Neale, B. M., Corvin, A., Walters, J. T., Farh, K. H., Holmans, P. A., ... & Pers, T. H. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature, 511(7510), 421.
作者:Ayden (brainnews创作团队成员)
校审/排版:Simon (brainnews编辑部)
,




