
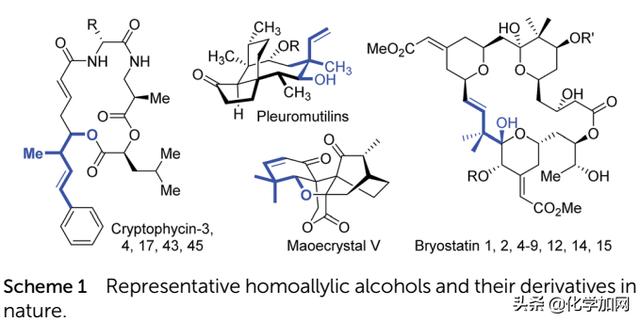
高烯丙基醇及其衍生物不仅是常见的合成中间体,还广泛存在于许多生物活性化合物和天然产物中(Scheme 1)。例如,cryptophycin家族产物显示出对实体瘤的突出活性,pleuromutilins抑制革兰氏阳性病原体的生长,maoecrystal V显示出对HeLa细胞系的潜在选择性,bryostatins对多种癌症和其他疾病具有显著的生物活性。因此,高烯丙基醇的催化合成一直是有机化学研究的热点之一。
(图片来源:Chem. Sci.)
过渡金属催化的酮/醛与1,3-二烯的还原烯丙基化是构建高烯丙基醇的有效方法。芳烃、二烯和羰基化合物之间的三组分串联反应可以实现复杂分子的快速组装,但相关研究依然有限。通过金属转移途径,化学家们已经实现了Ni-或Pt-催化的有机金属试剂与异戊二烯和醛的三组分加成(Fig. 1a)。从原子和步骤经济性来看,萜烯和羰基化合物上的定向C-H键加成是更具吸引力的方法。最近,Ellman和赵东兵等人分别报道了Co-和Rh-催化的二烯和醛上的C-H键加成。然而,当萜烯作为底物时,反应收率不理想且实例有限(Fig. 1b)。受上述研究的启发,四川大学周向葛课题组和中国科学院大连化学物理研究所陈庆安课题组团队设想开发一种萜烯与甲醛和芳烃之间的羟甲基化方法,该方法面临以下挑战:1)化学选择性控制:就亲电性而言,甲醛是比萜烯更好的偶联配偶体;2)区域选择性控制:萜烯具有至少两个类似的C=C键,与碳-金属络合物的迁移插入至少存在四种不同的取向(Fig. 1c)。近日,该团队实现了钴催化的萜烯与甲醛和芳烃之间的羟甲基化,该成果发表于近期Chem. Sci.(DOI: 10.1039/c9sc03747k)。

(图片来源:Chem. Sci.)
首先,作者用2-苯基吡啶、异戊二烯和多聚甲醛作为模型底物开展研究(Table 1),并在Cp*Co(CO)I2(5 mol%)和AgSbF6存在下以29%的收率得到高烯丙基醇4a。通过对添加剂的考察发现,羧酸盐和羧酸均能加速反应,可能是通过协同金属化-去质子化途径促进C-H活化。值得注意的是,将乙酸的量增加至40 mol%时,收率更好。通过筛选溶剂发现,1,4-二氧六环是最佳溶剂。

(图片来源:Chem. Sci.)
在确定了最佳反应条件后,作者考察了该反应的底物范围(Table 2)。在标准条件下,苯并喹啉可以几乎定量的收率得到高烯丙基醇4b;2-苯基嘧啶以及在吡啶上含吸电子或给电子取代基的底物均是合适的底物。随后,作者进一步考察了苯环上的取代基。氟取代的芳烃可以顺利进行反应并以良好的收率(>85%)得到相应的产物。给电子基团如甲氧基和甲基不受取代位置影响,均具有良好的耐受性。此外,该方法还可以扩展到羟基取代的芳烃和含吸电子取代基(如乙酰基、氯、溴和三氟甲基)的底物。值得注意的是,所有间位和对位取代的芳烃均得到单一的区域异构体,而且2-萘基衍生的芳烃可以定量转化为高烯丙基醇4s。

(图片来源:Chem. Sci.)
接下来,作者考察了萜烯的适用范围(Table 3)。异戊二烯单元由2增加到4(5a、5b、5c)对反应性没有显著影响;然而,罗勒烯(5a的异构体)与甲醛和芳烃之间的反应未得到目标产物。由于含9个异戊二烯单元的solanesene(5d)的溶解性差,使得目标产物的收率较低;天然萜类化合物myrcenol(5f)和farnesenol(5g)的反应收率良好(79-81%);源自叶绿素的phytadiene(5e)和双环二萜sclarene(5h)也适用于该转化。

(图片来源:Chem. Sci.)
为了验证该方法的实用性,作者进行了克级规模反应并以84%的收率得到羟甲基化产物6a(Fig. 2),其经m-CPBA氧化得到环氧化物9(54%),并伴有少量七元环产物10。值得注意的是,质子溶剂(DCM/H2O,1:1)可促进环氧化物9的开环;因此,利用HOTf(30 mol%)作为添加剂,可以由6a直接合成10(37%)。

(图片来源:Chem. Sci.)
为了探索该反应的机理,作者进行了一些对照实验(Fig. 3)。在标准条件下,作者利用1a /或D5-1a进行了动力学同位素效应(KIE)实验,并且平行反应和竞争反应的KIE值分别为2.8和2.0(eq 1),表明C-H键的断裂可能是决速步骤。在回收的起始原料中存在氢原子取代的产物,表明在钴催化下存在可逆的C-H活化过程(eq 2)。当用AcOD作为添加剂时,目标产物的收率为70%,其中氘代发生在甲基上(D1-4a),表明该过程中涉及相对较少的H-D交换(eq 3)。当甲醛不参与反应时,发生异戊二烯与1a的氧化偶联,但由于没有外部氧化剂使得反应收率低(eq 4)。在不存在异戊二烯的情况下,可以得到少量羟基化产物8;然而,在标准条件下,却未得到羟基化产物8(eq 5)。两种组分的偶联结果(eq 4和5)均表明,萜烯与甲醛对形成的芳基钴中间体的配位能力不同,可能是导致其具有化学选择性的主要原因。

(图片来源:Chem. Sci.)
为了更好地解释产生区域选择性的原因,作者详细介绍了生成区域特异性高烯丙醇产物的潜在途径(Fig. 4)。基于不同的加成取向,将五元钴络合物A(生成于钴预催化剂和1a之间的C-H活化步骤)与异戊二烯加成可以产生四种加成模式(B1、B2、B3和B4)。然而,由于空间位阻(B1和B2)或β-消除问题(B3),1,2-/2,1-或3,4-加成产物基本不会产生。相反,4,3-加成产物B4可以通过β-消除产生C;然后,Co-H 键1,4-插入至异戊二烯单元中得到中间体D;最后,D与甲醛通过"椅式"过渡态(E)定向加成得到高烯丙基醇产物4a,并再生钴催化剂。

(图片来源:Chem. Sci.)
总结:川大周向葛课题组和大连化物所陈庆安课题组团队开发了一种钴催化的萜烯与多聚甲醛和芳烃之间的羟甲基化方法,其化学选择性主要是萜烯和甲醛之间的配位能力差异引起的,并且异戊二烯和导向基团的空间位阻和配位能力决定了区域选择性。此外,通过C-H加成途径,这种萜烯的双官能团化还具有高原子经济性和步骤经济性。
,




